Lipophilicity and ADMET: A Comprehensive Guide for Optimizing Drug Properties
Lipophilicity, quantified as LogP and LogD, is a fundamental physicochemical property that critically influences the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) of drug candidates.
Lipophilicity and ADMET: A Comprehensive Guide for Optimizing Drug Properties
Abstract
Lipophilicity, quantified as LogP and LogD, is a fundamental physicochemical property that critically influences the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) of drug candidates. This article provides a comprehensive overview for researchers and drug development professionals, exploring the foundational principles that link lipophilicity to key pharmacokinetic behaviors. It delves into established and emerging methodologies for measuring and predicting lipophilicity, addresses common challenges in optimization—such as balancing solubility with permeability and mitigating toxicity risks—and validates strategies through case studies and comparative analysis of computational tools. By synthesizing current research and practical applications, this guide aims to support the rational design of compounds with superior ADMET profiles.
The Fundamental Role of Lipophilicity in Governing ADMET Properties
Lipophilicity represents one of the most fundamental physicochemical properties in drug discovery and development, serving as a critical determinant of a compound's behavior in biological systems. Defined as the affinity of a molecule for a lipophilic environment versus an aqueous one, lipophilicity directly influences a compound's absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile [1]. Within pharmaceutical sciences, lipophilicity is quantitatively described by two primary parameters: the partition coefficient (LogP) and the distribution coefficient (LogD) [2]. These descriptors enable researchers to predict how drug candidates will navigate the complex physiological environments within the human body, from gastrointestinal absorption to blood-brain barrier penetration [3]. A comprehensive understanding of LogP and LogD, their theoretical foundations, measurement techniques, and impact on drug disposition provides an essential framework for optimizing candidate compounds and reducing attrition in later development stages.
Fundamental Concepts: LogP and LogD
LogP: The Partition Coefficient
The partition coefficient (LogP) quantifies the equilibrium distribution of a single, unionized chemical species between two immiscible phases, typically n-octanol and water [2]. LogP is mathematically defined as the base-10 logarithm of the ratio of the compound's concentration in the organic phase to its concentration in the aqueous phase:
where [Drug]ₒcₜₐₙₒₗ represents the concentration of the unionized drug in octanol and [Drug]wₐₜₑᵣ represents the concentration in water [4]. This parameter provides a pure measure of intrinsic lipophilicity, considering only the neutral form of the molecule [2]. LogP values typically range from -2 to 7, with higher values indicating greater lipophilicity [5].
LogD: The Distribution Coefficient
The distribution coefficient (LogD) extends the concept of LogP by accounting for all forms of a compound present at a specific pH, including ionized, partially ionized, and unionized species [2]. LogD is defined as:
where [Ion]wₐₜₑᵣ represents the concentration of ionized drug species in the aqueous phase [4]. Unlike LogP, which is a constant for a given compound, LogD is pH-dependent and provides a more accurate representation of a drug's lipophilicity under physiologically relevant conditions [2].
Table 1: Key Differences Between LogP and LogD
| Parameter | LogP | LogD |
|---|---|---|
| Definition | Partition coefficient for unionized species only | Distribution coefficient accounting for all species |
| pH Dependence | pH-independent | pH-dependent |
| Ionization Consideration | No | Yes |
| Biological Relevance | Limited, as it doesn't reflect physiological conditions | High, as it models specific biological environments |
| Measurement Complexity | Simpler | More complex due to pH variability |
Theoretical Relationship Between LogP, LogD, and pKa
For ionizable compounds, a direct mathematical relationship exists between LogP, LogD, and the acid dissociation constant (pKa). This relationship allows for the calculation of LogD at any given pH based on the compound's intrinsic lipophilicity (LogP) and ionization properties:
- For basic compounds: LogD = LogP - log₁₀(1 + 10^(pH - pKa))
- For acidic compounds: LogD = LogP - log₁₀(1 + 10^(pKa - pH))
These equations demonstrate how ionization dramatically affects observed lipophilicity, particularly when the environmental pH approaches the compound's pKa value [4].
Experimental Determination Methods
shake-Flask Method
The traditional shake-flask method remains a gold standard for experimental LogP/LogD determination. This direct approach involves dissolving the compound in a pre-saturated mixture of n-octanol and water or buffer, vigorously shaking to facilitate partitioning, allowing phases to separate, and quantitatively analyzing the solute concentration in each phase using techniques such as UV spectrophotometry or HPLC [4]. While highly accurate, this method is time-consuming and requires relatively pure compounds, making it less suitable for high-throughput screening.
Chromatographic Methods
Reverse-phase thin-layer chromatography (RP-TLC) and high-performance liquid chromatography (HPLC) offer indirect, high-throughput alternatives for lipophilicity assessment. In RP-TLC, the retention factor (Rₘ) is measured and correlated with LogP/LogD values using standard compounds with known lipophilicity [5]. HPLC methods utilize retention times on reverse-phase columns to estimate lipophilicity, with the logarithm of the capacity factor (log k) correlating with LogP/LogD. Chromatographic approaches are particularly valuable for impure samples or compound mixtures and can be automated for higher throughput.
Table 2: Comparison of Experimental Methods for Lipophilicity Determination
| Method | Principle | Throughput | Advantages | Limitations |
|---|---|---|---|---|
| Shake-Flask | Direct partitioning between octanol/water | Low | High accuracy; reference method | Time-consuming; requires pure compounds |
| RP-TLC | Correlation of retention factor with lipophilicity | Medium | Suitable for impure samples; cost-effective | Indirect measurement; requires calibration |
| HPLC | Correlation of retention time with lipophilicity | High | Automation potential; precise | Method development required; instrumental complexity |
Research Reagent Solutions
Table 3: Essential Materials for Lipophilicity Assessment
| Reagent/Equipment | Function/Application |
|---|---|
| n-Octanol | Organic solvent simulating lipid membranes in partition studies |
| Buffer Solutions | Maintaining specific pH conditions for LogD measurements |
| Reverse-Phase TLC Plates | Stationary phase for chromatographic lipophilicity assessment |
| C18 HPLC Columns | Stationary phase for high-performance chromatographic methods |
| UV-Vis Spectrophotometer | Quantifying compound concentration in shake-flask experiments |
| pH Meter | Precise measurement and adjustment of aqueous phase pH |
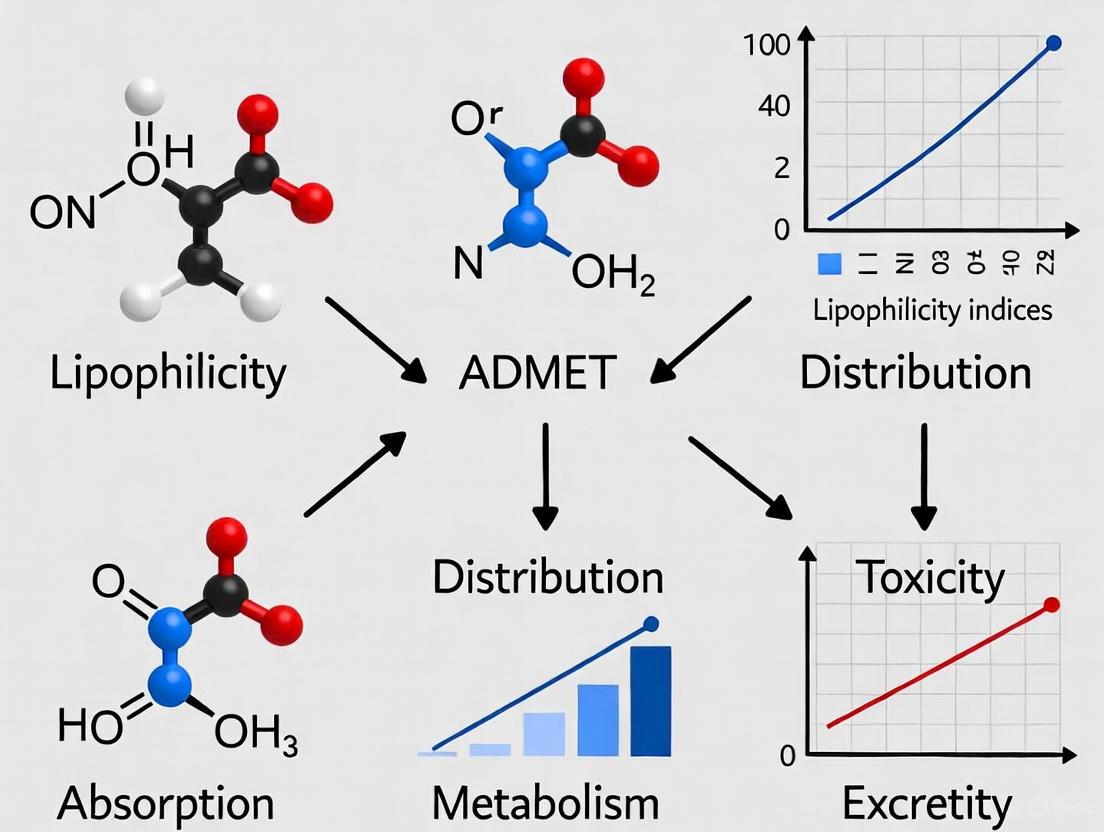
Impact of Lipophilicity on ADMET Properties
Lipophilicity serves as a master variable influencing virtually all aspects of a drug's disposition and action. The relationship between lipophilicity and key ADMET parameters follows often predictable patterns that enable medicinal chemists to optimize compound properties.
Absorption and Distribution
Lipophilicity fundamentally governs a compound's ability to cross biological membranes through passive diffusion. Optimal LogP values for oral absorption typically range between 1 and 5, balancing sufficient hydrophilicity for dissolution in gastrointestinal fluids with adequate lipophilicity for membrane permeation [4]. Highly lipophilic compounds (LogP > 5) often suffer from poor aqueous solubility, limiting their absorption, while excessively hydrophilic compounds (LogP < 0) may struggle to traverse lipid bilayers [2] [5].
For distribution, lipophilicity influences tissue penetration, volume of distribution, and plasma protein binding. Compounds with moderate lipophilicity (LogP ~2) demonstrate optimal blood-brain barrier penetration, while highly lipophilic drugs tend to accumulate in adipose tissue and exhibit increased protein binding, reducing their free concentration available for pharmacological activity [5] [3].
Metabolism and Excretion
Lipophilicity directly affects metabolic susceptibility, with increasingly lipophilic compounds generally undergoing more extensive metabolism by cytochrome P450 enzymes [3]. This relationship stems from both the substrate specificity of metabolic enzymes and the accessibility of lipophilic compounds to enzyme active sites located within endoplasmic reticulum membranes. For excretion, hydrophilic compounds are preferentially eliminated via renal clearance, while lipophilic compounds require metabolic conversion to more hydrophilic metabolites before renal or biliary excretion [3].
Toxicity Considerations
Excessive lipophilicity (LogP > 5) correlates with several toxicity risks, including phospholipidosis, promiscuous enzyme inhibition, and hERG channel binding associated with cardiotoxicity [5]. These associations have established lipophilicity control as a crucial strategy in mitigating attrition due to safety concerns in drug development.
The following diagram illustrates the multifaceted relationship between lipophilicity and key ADMET properties:
Computational Prediction Approaches
With the expansion of chemical space exploration in drug discovery, computational methods for predicting LogP and LogD have become indispensable tools for prioritizing compounds. These approaches range from fragment-based methods that calculate lipophilicity by summing contributions from molecular substructures to machine learning models trained on large experimental datasets [6].
Several software platforms offer advanced prediction capabilities, with recent benchmarks demonstrating high accuracy in blind challenges. For instance, Chemaxon's logP predictor achieved the lowest root mean square error (RMSE) in the SAMPL6 challenge, with only one structure exhibiting greater than 0.5 log unit deviation from experimental values [6]. Similarly, their pKa prediction algorithm demonstrated superior performance in the SAMPL7 blind challenge, enabling accurate LogD calculation across pH ranges [6].
These computational tools allow researchers to screen virtual compound libraries efficiently, optimize lead compounds through structural modification, and predict behavior under various physiological pH conditions before committing resources to synthesis and experimental testing.
LogP and LogD represent complementary descriptors that provide essential insights into a compound's physicochemical behavior and biological fate. While LogP characterizes intrinsic lipophilicity of the neutral form, LogD offers a more physiologically relevant perspective by accounting for ionization states at specific pH values. The experimental determination of these parameters through shake-flask, chromatographic, and other methods provides critical data for understanding compound behavior, while computational approaches enable high-throughput prediction and optimization.
Within the context of ADMET research, lipophilicity serves as a central parameter influencing absorption, distribution, metabolism, excretion, and toxicity. The optimal lipophilicity range for drug candidates represents a balance between opposing factors—sufficient hydrophilicity for dissolution and sufficient lipophilicity for membrane permeation. As drug discovery increasingly ventures beyond traditional chemical space, particularly with macrocycles, protein-based agents, and other modalities beyond the Rule of 5, understanding and controlling lipophilicity through both LogP and LogD remains crucial for designing compounds with favorable ADMET profiles and ultimately reducing attrition in drug development.
Lipophilicity as a Master Parameter in Drug Disposition and Pharmacokinetics
Lipophilicity, quantitatively expressed as the partition coefficient (LogP) or distribution coefficient (LogD), stands as a fundamental physicochemical property that profoundly influences the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of drug candidates. Its role as a master parameter stems from its direct control over passive drug permeation across biological membranes, solubility, and nonspecific binding to proteins and tissues. This whitepaper examines the central role of lipophilicity in drug design, summarizing its quantitative relationships with key pharmacokinetic and safety outcomes. Furthermore, it provides a detailed overview of established protocols for its experimental determination and computational prediction, serving as a technical guide for researchers and drug development professionals engaged in optimizing the pharmacokinetic profiles of new chemical entities.
Lipophilicity is a key physicochemical parameter that has to be widely taken into account when developing new drugs since it has been reported to have a significant influence on various pharmacokinetic properties such as absorption, distribution, permeability, and the routes of drug clearance [7]. Physically, lipophilicity is described as the logarithmic n-octanol-water partition coefficient (logP) that is characteristic of a given chemical [5]. This parameter has been extensively used in studies on the quantitative relationship between the structure and the activity (QSAR) [5].
The optimization of pharmacokinetic properties remains one of the most challenging aspects of drug design, and lipophilicity serves as a critical master parameter in this process [8]. Key pharmacokinetic parameters, clearance and volume of distribution, are multifactorial, which makes deriving structure-pharmacokinetic relationships difficult [8]. Lipophilicity influences the pharmacodynamic and toxicological profiles of compounds, affecting their ability to bind to plasma proteins and to interact with receptors at the drug's target site of action [5] [9]. Consequently, accurate determination and intelligent modulation of lipophilicity are essential for designing effective and safe pharmaceutical agents.
Core Concepts and Definitions
Lipophilicity Parameters: LogP vs. LogD
- LogP: The partition coefficient (P) is defined as the ratio of the equilibrium concentrations of a neutral compound in a two-phase system consisting of two immiscible solvents, typically n-octanol and water. LogP is its decimal logarithm and describes the intrinsic lipophilicity of a compound in its un-ionized form [7].
- LogD: The distribution coefficient (D) accounts for the ionization of compounds at a specific pH (commonly pH 7.4, noted as LogD7.4). It represents the ratio of the sum of the concentrations of all forms of the compound (ionized plus un-ionized) in the organic phase to the sum of the concentrations of all forms in the aqueous phase [10]. For compounds with low basicity, such as some pyridine derivatives, no difference between LogD7.4 and LogP values may be observed [10].
The Interplay with Acid-Base Properties (pKa)
The pKa of a drug directly influences its ionization state, which is intrinsically correlated to lipophilicity, solubility, affinity to proteins, and permeability across membranes [6]. The majority of drugs are weak acids and/or bases, and their ionization state changes across different physiological pH environments, from the highly acidic stomach (pH ~2) to the physiological pH of blood and tissues (pH 7.4) and the weakly acidic lysosomes (pH ~5) [6]. This relationship is crucial for understanding a drug's behavior in vivo, as only the un-ionized form of a drug can passively diffuse through lipid membranes.
Experimental and Computational Assessment of Lipophilicity
Experimental Methodologies
Shake-Flask Method
The classic shake-flask procedure, recommended by the Organization for Economic Co-operation and Development, involves the direct measurement of the partition coefficient [11]. A compound is partitioned between n-octanol and a buffer (e.g., TRIS buffer, pH 7.4), the phases are separated after equilibration, and the concentration of the compound in each phase is quantified (e.g., by UV spectroscopy). While considered a gold standard, this method is time-consuming, requires relatively large amounts of pure compounds, and is generally suitable for LogP values in the range of -2 to 4 [11].
Chromatographic Methods
Chromatographic techniques are widely used indirect methods to determine lipophilicity parameters experimentally.
- Reversed-Phase Thin-Layer Chromatography (RP-TLC): This method uses modified silica gel (e.g., RP-18) as the stationary phase and a water-organic mixture (e.g., acetone-TRIS buffer) as the mobile phase [9] [11]. The retention factor (Rf) is measured, and the derived parameter RM0 is calculated by extrapolating the RM value (RM = log(1/Rf - 1)) to zero concentration of the organic modifier. RM0 is considered a chromatographic lipophilicity index [9].
- Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC): This method determines lipophilicity through the retention time, often expressed as logk0. It offers high reproducibility and requires a smaller amount of sample compared to the shake-flask method [11].
A comparison of lipophilicity measurement techniques is provided in the table below.
Table 1: Comparison of Key Experimental Methods for Lipophilicity Determination
| Method | Principle | Lipophilicity Index | Advantages | Limitations |
|---|---|---|---|---|
| Shake-Flask [11] | Direct partitioning between n-octanol and aqueous buffer | LogP / LogD | Considered a gold standard; direct measurement | Time-consuming; requires pure compounds; limited logP range (-2 to 4) |
| RP-TLC [9] [11] | Retention on a non-polar stationary phase | RM0, φ0 | Low solvent consumption; high throughput; small sample amount | Indirect measurement; result can be system-dependent |
| RP-HPLC [11] | Retention on a non-polar stationary phase | logk0 | High accuracy and reproducibility; small sample amount | Indirect measurement; requires specialized equipment |
| 19F NMR [10] | Partitioning measured via 19F NMR signal integration in each phase | LogD7.4 | Useful for fluorinated compounds; internal standard can be used | Limited to compounds containing fluorine or other NMR-active nuclei |
Specialized Techniques: 19F NMR for Fluorinated Compounds
For fluorinated compounds, LogD7.4 can be determined using a variation of the shake-flask method combined with 19F NMR spectroscopy [10]. Together with a fluorinated internal standard of known lipophilicity, the compound is partitioned between octanol and an aqueous buffer (e.g., pH 7.4). Simple 19F NMR experiments allow for the calculation of the LogD7.4 value through the integration of NMR signals in each phase [10].
Computational Prediction (In Silico Methods)
A persistent need for accurate in silico models for lipophilicity prediction has arisen to facilitate the drug design process [7]. Numerous computational programs based on various mathematical algorithms are available for the online prediction of LogP.
Table 2: Commonly Used Software and Algorithms for Predicting Lipophilicity
| Software/Algorithm | Description | Availability |
|---|---|---|
| CLOGP | A widely used program based on fragment contributions [7] | Commercial |
| ALOGPs | An online tool that is part of the VCCLAB server [5] | Web-based |
| iLOGP | A method available on the SwissADME server [5] [11] | Web-based |
| XLOGP2/3 | Topology-based methods available on various servers [5] [9] | Web-based |
| MLOGP | A method based on molecular topology and Moriguchi parameters [5] [9] | Web-based / SwissADME |
| SILICOS-IT | A filter-based method for property prediction [5] [9] | Web-based / SwissADME |
| Chemaxon logP | A machine-learning-based model that performed well in blind challenges [6] | Commercial |
The calculated logP value for a given compound can differ significantly depending on the algorithm used [5] [9]. Therefore, it is considered good practice to use several prediction tools and to complement in silico data with experimental measurements, especially during the later stages of drug development [11].
The following diagram illustrates the typical workflow for assessing lipophilicity during drug discovery.
Lipophilicity Assessment Workflow
Lipophilicity as a Determinant of ADMET Properties
Absorption and Permeability
Lipophilicity plays a significant role in the transport of molecules across membranes, including the intestinal epithelium, the blood-brain barrier (BBB), and the skin [5] [12]. It is a key property in passive drug permeation processes [12]. Compounds with moderate lipophilicity tend to be better absorbed through cell membranes [11]. However, excessively high lipophilicity (LogP > 5) is associated with poor aqueous solubility, which can limit absorption from the gastrointestinal tract [5] [7]. The ability to cross the BBB is also influenced by lipophilicity; high lipophilicity can promote nonspecific binding to plasma proteins, potentially reducing the amount of free drug available to penetrate the BBB [5].
Distribution and Metabolism
Lipophilic substances can more easily penetrate cell membranes and migrate to lipid-rich tissues, which affects their distribution in the body and can lead to a larger volume of distribution [11]. Substances with increased lipophilicity may be more susceptible to metabolism in the liver through oxidation, reduction, and conjugation reactions, leading to a fast metabolic turnover [5] [11]. This can impact the pharmacological activity, duration of action, and potential toxicity of a drug.
Toxicity and Safety Liabilities
The lipophilicity of drugs is closely related to their toxicity, as it can affect their accumulation in tissues and interactions with receptors and proteins in the body [13] [11]. Highly lipophilic compounds have an increased risk of being promiscuous binders, leading to off-target effects [6]. The "3/75 rule" (ClogP < 3 and TPSA > 75 Ų), proposed by Pfizer, suggests that drug candidates in this chemical space are considerably less likely to cause significant toxicological effects at total plasma concentrations below 10 μM [13]. An analysis of Takeda internal compounds supported this trend, showing that reducing lipophilicity noticeably decreases toxicity odds [13]. The relationship between lipophilicity and toxicity is also influenced by the ionization state of the molecule, with basic molecules showing a different toxicity odds profile compared to neutral or acidic molecules [13].
Table 3: Impact of Lipophilicity on Key ADMET Properties
| ADMET Property | Impact of Low Lipophilicity | Impact of High Lipophilicity | Optimal Range/Considerations |
|---|---|---|---|
| Absorption / Permeability [5] [12] [11] | Poor passive permeation through lipid membranes | Good membrane permeation, but potential solubility-limited absorption | Moderate lipophilicity (e.g., ~LogP 2) often shows optimal abilities to reach targets [5] |
| Solubility [7] [6] | Generally high aqueous solubility | Poor aqueous solubility; risk of precipitation | Balance needed for adequate dissolution and absorption |
| Distribution [11] [7] | Low volume of distribution; limited tissue penetration | High volume of distribution; potential accumulation in fatty tissues | Influenced by ionization state; basics tend to have higher Vd [13] |
| Metabolism [5] [11] | Slower metabolic turnover | Fast metabolic turnover (Phase I oxidation) | High lipophilicity increases susceptibility to liver metabolism |
| Toxicity [5] [13] [6] | Lower risk of promiscuity and tissue accumulation | Increased risk of promiscuous binding, off-target effects, and tissue accumulation | "3/75 rule" (ClogP < 3, TPSA > 75) associated with lower toxicity odds [13] |
| Protein Binding [5] [13] | Generally lower plasma protein binding | High plasma protein binding, reducing free drug concentration | Acidic compounds tend to have higher plasma protein binding [13] |
The following diagram summarizes the multifaceted relationship between lipophilicity and key ADMET outcomes.
Lipophilicity-ADMET Relationships
The Scientist's Toolkit: Key Reagents and Materials
Table 4: Essential Research Reagent Solutions for Lipophilicity Studies
| Reagent / Material | Function / Application | Example Use in Protocol |
|---|---|---|
| n-Octanol | Organic solvent simulating lipid environments in partition experiments [10] [7] | Shake-flask method; saturating aqueous buffers for LogP/LogD determination. |
| TRIS Buffer (pH 7.4) | Aqueous phase mimicking physiological pH in partition experiments [9] [11] | Used as aqueous phase in RP-TLC mobile phases and shake-flask methods. |
| RP-18 TLC/HPLC Plates | Stationary phase with bonded C18 chains for reversed-phase chromatography [9] [11] | Solid support for chromatographic determination of lipophilicity (RM0). |
| Acetone / Methanol | Organic modifiers for mobile phases in chromatographic methods [9] [11] | Used in gradient or isocratic elution to modulate retention in RP-TLC/RP-HPLC. |
| Deuterated Solvent (e.g., D₂O) | Solvent for NMR spectroscopy allowing for field locking | Used in 19F NMR-based LogD7.4 determination methods [10]. |
| Fluorinated Internal Standard | Reference compound with known LogP for quantitative NMR [10] | Added to partition experiments for 19F NMR-based LogD7.4 calculation. |
Lipophilicity is unequivocally a master parameter in drug disposition and pharmacokinetics, exerting a profound and multifaceted influence on every aspect of a drug's ADMET profile. A comprehensive understanding of its role enables drug developers to make more informed decisions during the design and optimization of new chemical entities. The optimal lipophilicity for a drug candidate is a delicate balance, as it must be sufficiently lipophilic to permeate membranes and reach its target, yet not so lipophilic that it incurs poor solubility, rapid metabolism, promiscuous binding, or toxicity. The integration of robust computational predictions with validated experimental methodologies, such as chromatographic techniques and the shake-flask method, provides a powerful strategy for navigating this balance. By systematically applying the principles and protocols outlined in this whitepaper, researchers can effectively prioritize compounds with a higher probability of success, thereby increasing the efficiency of the drug discovery process and contributing to the development of safer and more effective therapeutics.
Lipophilicity, quantitatively expressed as the partition coefficient (log P) or distribution coefficient (log D), is a fundamental physicochemical parameter that measures a molecule's affinity for lipid versus aqueous environments. It is defined as the ratio of the concentrations of a neutral compound in n-octanol and water phases under equilibrium conditions (log P = log Co/Cw) [14]. For ionizable compounds, the distribution coefficient (log D) accounts for all forms of the compound at a specific pH, making it more relevant for physiological conditions [6] [14]. In drug discovery, lipophilicity serves as a master variable that critically influences absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, ultimately determining a compound's therapeutic efficacy and safety profile [6] [15] [14].
The "Goldilocks Principle" in medicinal chemistry describes the essential balance required for lipophilicity—values that are too low result in poor membrane permeability and inadequate tissue penetration, while excessively high values lead to insufficient aqueous solubility, increased metabolic clearance, and higher risk of toxicity [6] [14]. This principle is operationalized through Lipinski's Rule of Five, which establishes that for optimal oral bioavailability, a compound's log P value should typically be below 5 [16] [14]. Analysis of approved drugs reveals that approximately 90% have log P values between 0 and 3, with an ideal range of 1-3 for balancing solubility and permeability requirements [14]. For specific therapeutic goals such as central nervous system penetration, the ideal log P value is approximately 2 [14]. Maintaining lipophilicity within this optimal range is crucial for mitigating late-stage clinical failures, with poor solubility being implicated in nearly 40% of such failures [17].
Quantitative Foundations: Key Lipophilicity Parameters and Their Measurements
Experimental Methodologies for Lipophilicity Determination
Accurate determination of lipophilicity employs both traditional and chromatographic techniques, each with distinct advantages and limitations. The shake-flask method, considered the gold standard, involves direct measurement of a compound's distribution between n-octanol and water phases under controlled conditions [15] [14]. While providing fundamental validation data, this approach is time-consuming and requires relatively large amounts of pure compounds, with a reliable measurement range of log P between -2 to 4 [15]. Modern chromatographic techniques have emerged as efficient alternatives that provide excellent correlation with traditional methods while offering enhanced throughput and reproducibility.
Table 1: Comparison of Experimental Methods for Lipophilicity Determination
| Method | Principle | log P Range | Advantages | Limitations |
|---|---|---|---|---|
| Shake-Flask [15] [14] | Direct partitioning between n-octanol/water | -2 to 4 | Gold standard, OECD-approved | Time-consuming, requires pure compounds |
| RP-TLC [15] [16] [18] | Retention on C18/C8 plates with aqueous-organic mobile phases | Wide range | High throughput, minimal solvent use | Multiple experimental variables |
| RP-HPLC [15] [16] [14] | Retention on C18 columns with methanol/water or acetonitrile/water gradients | Broad applicability | Excellent reproducibility, automated | Equipment cost, method development |
| Micellar Liquid Chromatography [14] | Use of biomimetic micellar mobile phases | NA | Better simulation of biological membranes | Less established protocols |
Chromatographic techniques, particularly Reversed-Phase High Performance Liquid Chromatography (RP-HPLC) and Reversed-Phase Thin Layer Chromatography (RP-TLC), have gained widespread acceptance for lipophilicity screening [15] [16] [18]. The Organization for Economic Co-operation and Development (OECD) endorses RP-HPLC as a preferred method, especially for compounds challenging to measure via shake-flask techniques [14]. These methods establish relationships between retention parameters (RM0 for TLC, log k0 for HPLC) and lipophilicity through linear solvent-strength models, enabling high-throughput determination while consuming minimal amounts of compound [15] [16]. RP-TLC offers additional advantages as a green analytical chemistry approach due to miniaturization and reduced solvent consumption [14]. Studies on diquinothiazines and pseudothiohydantoin derivatives have demonstrated excellent correlation between chromatographically determined lipophilicity parameters and computational predictions [15] [16].
Computational Approaches for Lipophilicity Prediction
In silico methods for lipophilicity prediction provide rapid screening capabilities essential for early-stage drug design. Multiple algorithms have been developed, each employing distinct mathematical approaches to estimate log P values:
Table 2: Computational Methods for Lipophilicity Prediction
| Method/Software | Algorithm Type | Performance Notes | Applications |
|---|---|---|---|
| iLOGP [15] | Structure-based | High similarity to chromatographic RM0 for certain diquinothiazines | Early-stage compound prioritization |
| XLOGP3 [15] [18] | Atom-based | Widely used in commercial packages | Drug-likeness screening |
| WLOGP [15] [18] | Fragmental | Good performance on diverse chemical spaces | Lead optimization |
| Chemaxon [6] | Hybrid | Top performer in SAMPL6 blind challenge (lowest RMSE) | Industrial drug design |
| mlogP [15] [18] | Molecular topology | Fast calculation for high-throughput screening | Virtual library design |
| SILCOS-IT [15] | Fragment-based | Good performance on complex heterocycles | Specialty chemical applications |
Computational tools such as SwissADME, pkCSM, and ADMET Predictor integrate multiple algorithms to provide comprehensive ADMET profiling alongside lipophilicity predictions [19] [15] [16]. Evaluation studies demonstrate that these tools achieve varying degrees of accuracy, with correlation coefficients (R²) for metabolic stability predictions ranging from 0.53 for global models to 0.72 for locally-trained models [19]. The development of large-scale benchmarks like PharmaBench, which comprises 156,618 raw entries from 14,401 bioassays, addresses previous limitations in dataset size and chemical diversity, enabling more robust model training and validation [20].
The Molecular Toolkit: Essential Reagents and Platforms for Lipophilicity Research
Table 3: Essential Research Reagents and Platforms for Lipophilicity and ADMET Studies
| Category | Specific Tools/Reagents | Function/Application | Key Features |
|---|---|---|---|
| Chromatographic Systems [15] [16] [18] | RP-18F254, RP-8F254, RP-2F254 TLC plates; C18 HPLC columns | Experimental lipophilicity determination | Reproducible retention parameters (RM0, log kw) |
| Organic Modifiers [15] [16] [18] | Acetone, acetonitrile, methanol, 1,4-dioxane | Mobile phase composition | Different selectivity and hydrogen-bonding capacity |
| Computational Platforms [19] [15] [16] | SwissADME, pkCSM, ADMET Predictor, PreADMET | In silico ADMET profiling | Multi-parameter optimization, high-throughput |
| LogP Prediction Algorithms [6] [15] [18] | iLOGP, XLOGP3, WLOGP, Chemaxon | Computational logP estimation | Various statistical approaches and training sets |
| Buffers and pH Control [15] | TRIS buffer (pH 7.4), phosphate buffers | Physiological simulation | Biorelevant conditions for log D determination |
| Reference Compounds [6] [18] | Standard drugs with known logP values | Method calibration and validation | Quality control and interlaboratory comparison |
Diagram 1: Integrated Lipophilicity Optimization Workflow in Drug Discovery
Lipophilicity in Practice: Case Studies and Experimental Protocols
Case Study 1: Diquinothiazine Anticancer Agents
A comprehensive investigation of fifteen dialkylaminoalkyldiquinothiazine hybrids with demonstrated anticancer activity exemplifies the practical application of lipophilicity-ADMET relationships [15]. Researchers employed both experimental (RP-TLC) and computational approaches to determine lipophilicity parameters and correlate them with pharmacokinetic profiles. The RP-TLC protocol utilized RP18F254 plates with acetone-TRIS buffer (pH 7.4) mobile phases to determine RM0 values, which were then compared against eight different computational algorithms (iLOGP, XLOGP3, WLOGP, MLOGP, SILCOS-IT, LogP, logP, and milogP) [15]. Results demonstrated that iLOGP showed the strongest correlation with chromatographic values for certain compounds, validating its utility for rapid prediction during early development stages. Subsequent in silico ADMET profiling using SwissADME and pkCSM platforms provided comprehensive pharmacokinetic data, enabling the identification of compounds with optimal lipophilicity-balanced profiles for further development as anticancer candidates [15].
Case Study 2: Pseudothiohydantoin Derivatives as 11β-HSD1 Inhibitors
A study of 28 pseudothiohydantoin derivatives with inhibitory activity toward 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) illustrates the critical role of lipophilicity in optimizing central nervous system-targeted therapeutics [16]. Researchers implemented a dual-chromatographic approach using both RP-TLC and RP-HPLC with methanol as the organic modifier to determine lipophilicity parameters (RM0 and log kw). The investigation revealed that 27 of the 28 tested compounds exhibited log P values < 5, complying with Lipinski's Rule of Five and indicating high potential for oral bioavailability [16]. Structural analysis demonstrated clear relationships between substituent characteristics and lipophilicity, with larger hydrophobic groups at the 2-position of the thiazole ring consistently increasing lipophilicity parameters. This systematic approach enabled the team to select candidates with optimal lipophilicity (log P ~2) for blood-brain barrier penetration while maintaining sufficient aqueous solubility for formulation and absorption [16].
Detailed Experimental Protocol: RP-TLC for Lipophilicity Screening
Materials and Equipment:
- Stationary Phase: HPTLC RP-18F254, RP-8F254, or RP-2F254 plates (Merck)
- Mobile Phase: Acetone, acetonitrile, or methanol with TRIS buffer (pH 7.4) in varying proportions (e.g., 40:60, 50:50, 60:40, 70:30, 80:20 organic:buffer)
- Sample Preparation: 1 mg/mL solutions in methanol, applied as 1 μL spots
- Detection: UV light at 254 nm or appropriate wavelength
- Chromatographic Chamber: Saturated twin-trough glass chamber
Procedure:
- Condition the TLC plates in the chromatographic chamber for 30 minutes
- Apply sample spots 1 cm from the bottom edge using capillary pipettes
- Develop chromatograms in mobile phases with increasing organic modifier concentration
- Dry plates and detect spots under UV light
- Measure retention factors (Rf) for each compound at different mobile phase compositions
- Calculate RM values using the equation: RM = log (1/Rf - 1)
- Plot RM values against organic modifier concentration (φ) and extrapolate to φ = 0 to obtain RM0
- Validate with standard compounds of known log P values
Data Interpretation: The RM0 parameter serves as the chromatographic lipophilicity index, with higher values indicating greater lipophilicity. For the diquinothiazine series, RM0 values ranged from 1.92 to 3.52, indicating moderate to high lipophilicity appropriate for membrane penetration while remaining within drug-like space [15]. This protocol enables rapid screening of 20-30 compounds per day with minimal material consumption (~100 μg per compound), making it ideal for early-stage prioritization [15] [18].
Diagram 2: Relationship Between Lipophilicity and ADMET Properties
The critical importance of maintaining optimal lipophilicity in drug design cannot be overstated. The Goldilocks Principle—finding the "just right" balance between hydrophilicity and lipophilicity—represents a fundamental concept in modern medicinal chemistry that directly addresses major causes of clinical-stage attrition [17] [14]. Through integrated approaches combining chromatographic experimentation and computational prediction, researchers can efficiently navigate chemical space to identify compounds with balanced ADMET profiles. The ideal lipophilicity range of log P 0-3 (approximately log P ~2 for CNS targets) emerges as a consistent theme across diverse therapeutic areas and chemical classes [15] [16] [14].
Future directions in lipophilicity optimization will likely focus on several key areas. First, the development and expansion of comprehensive benchmark sets like PharmaBench, which addresses previous limitations through large-scale data mining of 14,401 bioassays, will enable more accurate and chemically relevant predictive models [20]. Second, advanced machine learning approaches, including autoencoder-based latent space augmentation as demonstrated in LatMixSol for solubility prediction, offer promising strategies for addressing data scarcity and improving prediction accuracy for novel chemical entities [17]. Finally, the integration of multi-parameter optimization considering the emerging concept of "informacophores"—data-driven molecular representations that capture essential features for biological activity—will provide more holistic frameworks for compound design [21]. As these technologies mature, the systematic application of the Goldilocks Principle for lipophilicity optimization will continue to play a central role in accelerating the discovery of safer and more effective therapeutics.
{# Lipophilicity's Direct Impact on Absorption and Intestinal Permeability}
Lipophilicity, quantified as the partition coefficient (log P), is a fundamental physicochemical property that directly governs a drug molecule's passive diffusion across intestinal epithelial cells. While moderate lipophilicity enhances membrane permeability, evidence demonstrates that excessively high lipophilicity (log P > 3.5) paradoxically decreases intestinal transport due to membrane retention and poor aqueous solubility. This whitepaper examines the non-linear relationship between lipophilicity and intestinal absorption, details key experimental methodologies for its assessment, and contextualizes these findings within the critical framework of ADMET property optimization in drug design.
Within drug discovery, the ADMET properties—Absorption, Distribution, Metabolism, Excretion, and Toxicity—are pivotal to a compound's clinical success. Among these, intestinal absorption serves as the initial gateway for oral drugs. A compound's lipophilicity is a principal descriptor influencing its pharmacokinetic behavior [5] [15]. It determines the balance between a drug's solubility in the aqueous fluids of the gastrointestinal tract and its permeability through the lipophilic bilayers of intestinal cell membranes [6]. Understanding the direct and often non-linear impact of lipophilicity on intestinal permeability is therefore essential for the rational design of bioactive compounds with optimal oral bioavailability.
The Parabolic Relationship: An Optimal Lipophilicity Range
The conventional assumption that permeability increases monotonically with lipophilicity is an oversimplification. Research reveals a more nuanced, parabolic relationship.
Key Experimental Evidence
A seminal study using intestinal epithelial cell lines (HT29-18-C1 and Caco-2) demonstrated that the transepithelial permeability coefficient increases with the octanol/buffer distribution coefficient (log Do/b) only up to a value of approximately 3.5. For compounds with log Do/b values between 3.5 and 5.2, the permeability coefficient decreased with increasing lipophilicity [22]. This indicates that an octanol/buffer distribution coefficient near 3000 (log D ≈ 3.5) corresponds to an optimal transepithelial passage, and excessively lipophilic compounds exhibit low intestinal epithelial permeability and, consequently, low oral absorption [22].
The underlying mechanism for this phenomenon is membrane retention. Highly lipophilic drugs interact strongly with the hydrophobic tails of phospholipids in cell membranes. Instead of traversing the membrane, these compounds become sequestered within the lipid bilayer itself [23]. This is particularly critical for Biopharmaceutics Classification System (BCS) Class 2 (low solubility, high permeability) and Class 4 (low solubility, low permeability) drugs, where high lipophilicity exacerbates poor aqueous solubility, creating a double-edged sword for absorption [23].
Table 1: The Impact of Lipophilicity on Drug Properties and ADMET Profiles
| Lipophilicity (log P) | Impact on Intestinal Permeability | Associated ADMET Risks |
|---|---|---|
| Low ( < 2) | Low permeability due to poor membrane partitioning | Poor absorption, limited distribution [5] |
| Moderate (2 - 3.5) | Optimal permeability with minimal membrane retention | Good absorption, favorable bioavailability [22] [5] |
| High (> 3.5 to 5) | Decreased permeability due to membrane retention [22] [23] | Poor solubility, rapid metabolic turnover, tissue accumulation [5] |
| Very High (> 5) | Significantly low permeability and absorption | High plasma protein binding, promiscuous binding, toxicity risks [5] [6] |
Experimental Protocols for Assessing Permeability and Lipophilicity
Accurate assessment requires robust and relevant experimental models. The following methodologies are central to evaluating the permeability of lipophilic drugs.
In Vitro Permeability Assay Using a Freestanding Lipid Bilayer
This protocol, developed to quantify transport and membrane retention, uses a solvent-free planar lipid bilayer for high-fidelity results [23].
- Primary Materials: 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), Squalane, drug compounds, UV cuvette, UV-Vis spectrophotometer.
- Lipid Bilayer Formation: A stable, freestanding planar lipid bilayer is formed within a standard UV cuvette. The bilayer is created by the adhesion between a lipid monolayer at a planar oil-water interface and another monolayer at a droplet interface [23].
- Permeability Measurement: The water droplet, containing the solubilized lipophilic drug (often with a minimal amount of DMSO or methanol to aid solubility), serves as the donor compartment. The drug transports across the bilayer into the acceptor buffer phase due to the concentration gradient. The transport is tracked in real-time by measuring UV absorbance [23].
- Quantifying Membrane Retention: A comprehensive transport model is applied to the concentration data over time to estimate the fraction of drug molecules trapped in the lipid bilayer, which is reported as the membrane retention fraction [23].
Determination of Lipophilicity by Reversed-Phase Thin-Layer Chromatography (RP-TLC)
RP-TLC is a widely used chromatographic technique for the experimental determination of lipophilicity descriptors [5] [15].
- Stationary Phase: RP-18 (C18) silica plates.
- Mobile Phase: Acetone-TRIS buffer (pH 7.4) mixtures or other water-organic modifier systems.
- Experimental Procedure: The compounds are spotted on the TLC plate, which is then developed in a chromatographic chamber with the mobile phase. The retention factor (RM) is calculated from the compound's migration distance. The lipophilicity parameter (RM^0^) is derived by extrapolating the RM values to 0% organic modifier, providing a value that correlates well with the octanol-water partition coefficient (log P) [15].
- Advantages: This method requires a small amount of sample, is rapid and cost-effective, and provides results that are consistent with the shake-flask method [15].
Table 2: Essential Research Reagents and Materials for Key Experiments
| Research Reagent / Material | Function in Experiment |
|---|---|
| Caco-2 / HT29-18-C1 Cell Lines | Differentiated human intestinal epithelial cells used as an in vitro model of the intestinal barrier for permeability studies [22]. |
| 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) | A phospholipid used to create freestanding planar lipid bilayers and liposomes that mimic biological membranes [23]. |
| Squalane | A hydrocarbon oil used in the formation of solvent-free freestanding lipid bilayers in permeability assays [23]. |
| RP-18 (C18) TLC Plates | The reversed-phase stationary phase used in RP-TLC for the experimental determination of lipophilicity parameters (RM^0^) [15]. |
| n-Octanol and Buffer Solutions | The two phases used in the shake-flask method and as a reference system for calculating the partition coefficient (log P), the standard index of lipophilicity [22] [24]. |
| Span 60 (Sorbitan Monostearate) | A non-ionic surfactant used in industrial granulation procedures to enhance the permeability of poorly absorbed drugs (e.g., Acyclovir) [24]. |
Visualization of Concepts and Workflows
Lipophilicity-Permeability Relationship: This diagram illustrates the parabolic relationship between drug lipophilicity and intestinal permeability, highlighting the optimal range and consequences of deviation.
Lipid Bilayer Permeability Assay: This workflow outlines the key steps in an in vitro permeability assay using a freestanding lipid bilayer to measure the transport of lipophilic drugs.
The direct impact of lipophilicity on intestinal permeability is a cornerstone of ADMET research. The evidence clearly argues against the simplistic "more lipophilic is better" paradigm, establishing instead that an optimal range (log P ~2-3.5) exists for maximal permeability. Exceeding this range introduces significant liabilities, including membrane retention and poor solubility, which can severely compromise oral bioavailability. Modern drug development leverages this understanding through integrated approaches, using in silico predictions [5] [20] [6] to guide the design of novel compounds and sophisticated in vitro models [23] to accurately profile their permeability early in the pipeline. The ongoing creation of large, high-quality benchmark datasets like PharmaBench [20] is poised to further empower machine learning models, enhancing our ability to predict and optimize this critical ADMET property for future drug candidates.
In modern drug discovery, the influence of a compound's lipophilicity on its Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties is a critical area of research [5] [25]. Distribution, a key component of pharmacokinetics, determines how a drug disseminates throughout the body to reach its site of action [26]. This process is governed by three interconnected factors: plasma protein binding, tissue penetration, and volume of distribution. Lipophilicity, quantitatively expressed as the logarithm of the n-octanol-water partition coefficient (logP) or the distribution coefficient (logD) that accounts for ionization, serves as a primary molecular descriptor that profoundly influences these distribution parameters [27] [25]. For drug development professionals, understanding these relationships is essential for optimizing candidate compounds, predicting therapeutic efficacy, and avoiding potential toxicity issues. This technical guide provides an in-depth examination of how lipophilicity modulates drug distribution characteristics, supported by experimental data and methodological protocols relevant to preclinical research.
Core Concepts and Lipophilicity Fundamentals
Defining Lipophilicity and Its Molecular Determinants
Lipophilicity represents a compound's affinity for lipophilic (fat-like) environments relative to aqueous environments [25]. Physically, it is described as the logarithmic n-octanol-water partition coefficient (logP) for neutral compounds or the distribution coefficient (logD) for ionizable compounds, which considers the extent of ionization at a specific pH [27] [5]. Mathematically, logD provides a more accurate representation of lipophilicity under physiological conditions for the 95% of drugs that contain ionizable groups [27].
The strategic importance of lipophilicity in drug design stems from its direct correlation with numerous ADMET properties [25]. Compounds with moderate lipophilicity (typically logD ~1-3) generally demonstrate optimal balance between solubility and permeability, leading to better absorption and distribution profiles [25]. Excessive lipophilicity (logP > 5) correlates with undesirable properties including poor aqueous solubility, increased metabolic clearance, tissue accumulation, and strong plasma protein binding that reduces free drug concentration [5].
Quantitative Impact of Lipophilicity on Distribution Properties
Table 1: Correlation Between Lipophilicity and Key Distribution Parameters
| Lipophilicity Range (LogD₇.₄) | Plasma Protein Binding | Tissue Penetration Potential | Typical Volume of Distribution | Clinical Implications |
|---|---|---|---|---|
| <1 | Low | Low (hydrophilic) | Small (0.25 L/kg) | Renal clearance dominant; limited tissue distribution [25] |
| 1-3 | Moderate | Moderate | Moderate | Balanced distribution; optimal for many drug classes [25] |
| 3-5 | High | High | Large | Extensive tissue distribution; possible accumulation [25] |
| >5 | Very high | Very high but variable | Very large | Poor solubility; high metabolic clearance; tissue accumulation [5] [25] |
Plasma Protein Binding
Mechanisms and Significance
Plasma protein binding refers to the reversible association of drug molecules with proteins in the blood, primarily albumin (for acidic drugs) and α₁-acid glycoprotein (for basic drugs) [28]. This binding creates a reservoir of inactive drug that is in equilibrium with the free, pharmacologically active fraction [26] [29]. Only the unbound drug can diffuse across cellular membranes to reach target sites, undergo metabolism, or be excreted [28] [29].
The degree of plasma protein binding significantly influences a drug's distribution profile. Highly protein-bound drugs (>90%) typically exhibit longer half-lives as the bound fraction acts as a sustained-release reservoir, but may demonstrate limited tissue penetration due to reduced free fraction availability [26] [30]. For drugs with high binding (>95%), even small changes in binding affinity or protein concentration can lead to substantial increases in free drug concentration, potentially resulting in toxicity [28].
Lipophilicity-Protein Binding Relationship
Lipophilicity directly correlates with the extent of plasma protein binding [25]. More lipophilic compounds possess greater affinity for hydrophobic binding pockets on plasma proteins, particularly albumin [28]. This relationship follows a generally linear pattern across moderate lipophilicity ranges, though it may plateau at extreme logP values due to solubility limitations.
Table 2: Experimental Data on Lipophilicity, Protein Binding and Permeability
| Drug Compound | LogP | LogD₆.₈ | Buccal Permeability Kp (×10⁻⁶ cm/s) | Protein Binding (%) | Clinical Notes |
|---|---|---|---|---|---|
| Nimesulide | 1.94 | 1.69 | 30.0 ± 6.0 | Not specified | High permeability [27] |
| Verapamil | 3.79 | 1.72 | 25.1 ± 3.6 | Not specified | CYP3A4 substrate [26] [27] |
| Lidocaine | 2.10 | 1.20 | 17.0 ± 1.8 | Not specified | Moderate permeability [27] |
| Propranolol | 3.48 | 1.20 | 14.0 ± 1.7 | Not specified | High permeability [27] |
| Amitriptyline | 5.04 | 1.64 | 13.4 ± 1.8 | Not specified | Extensive distribution [27] |
| Diltiazem | 2.79 | 1.04 | 7.3 ± 0.7 | Not specified | Moderate permeability [27] |
| Caffeine | -0.07 | -0.07 | 9.0 ± 0.5 | Not specified | High permeability despite low logP [27] |
| Naproxen | 3.18 | 0.60 | 3.8 ± 0.3 | >99% | High protein binding [27] [29] |
| Warfarin | 2.60 | 0.70 | 1.6 ± 0.2 | 97-99% | High protein binding; narrow therapeutic index [27] [29] |
| Metoprolol | 1.95 | -0.56 | 1.3 ± 0.2 | Not specified | Low permeability [27] |
| Pindolol | 1.83 | -0.90 | 0.12 ± 0.01 | Not specified | Very low permeability [27] |
Methodological Approach: Plasma Protein Binding Assays
Equilibrium Dialysis Protocol (Adapted from Bardal et al. and Sciencedirect Topics) [28]:
Apparatus Setup: Utilize a two-chamber dialysis system separated by a semi-permeable membrane with a molecular weight cutoff that retains the protein while allowing free drug passage.
Sample Preparation:
- Prepare drug solution in phosphate buffer (pH 7.4) at therapeutic concentrations (typically 1-100 μM)
- Add human serum albumin or α₁-acid glycoprotein at physiological concentrations (35-40 g/L for albumin, 0.4-1 g/L for α₁-acid glycoprotein)
- Maintain temperature at 37°C with continuous gentle agitation
Equilibrium Establishment:
- Allow system to reach equilibrium (typically 4-24 hours depending on drug properties)
- Confirm equilibrium by sampling both chambers at multiple timepoints
Analysis:
- Sample both protein-containing and buffer chambers
- Quantify drug concentrations using HPLC-UV, LC-MS/MS, or scintillation counting for radiolabeled compounds
- Calculate fraction unbound (fᵤ) = Cbuffer/Cprotein chamber
- Percent bound = (1 - fᵤ) × 100
Data Interpretation:
- For drugs with concentration-independent binding, fᵤ remains constant across therapeutic range
- For drugs with capacity-limited binding, monitor for concentration-dependent changes in fᵤ
Tissue Penetration
Mechanisms of Tissue Penetration
Tissue penetration describes a drug's ability to cross biological membranes and distribute into various tissues and organs [30]. The rate and extent of penetration depend on the drug's physicochemical properties and the physiological characteristics of the target tissue [26]. Lipophilic drugs primarily cross membranes via passive diffusion through the lipid bilayer, while hydrophilic compounds may utilize paracellular pathways or active transport mechanisms [27] [31].
Specialized barriers like the blood-brain barrier (BBB) significantly restrict tissue penetration based on a drug's physicochemical properties [26] [25]. The BBB favors lipophilic compounds with molecular weight <500 Da, while effectively excluding large, hydrophilic molecules [26]. ΔlogP, a measure of the difference between partition coefficients in different solvent systems, has been used as an indicator for blood-brain partitioning, with higher ΔlogP values correlating with lower brain-to-blood ratios [25].
Lipophilicity-Permeability Relationship
Lipophilicity fundamentally governs a drug's permeability across biological membranes [27] [25]. The relationship between logD and permeability typically follows a sigmoidal pattern, with permeability increasing with lipophilicity up to an optimal range, beyond which it may plateau or decrease due to factors like unstirred water layer effects or membrane retention [27].
Research on buccal permeability demonstrated that logD provides a better correlation with permeability than logP for ionizable drugs, as it accounts for the ionization state at physiological pH [27]. In this study, drugs with logD₆.₈ > 1.0 (nimesulide, verapamil, lidocaine) showed significantly higher permeability coefficients compared to those with logD₆.₈ < 0 (metoprolol, pindolol) [27].
Figure 1: Lipophilicity influences distribution through multiple interconnected pathways. Enhanced lipophilicity improves passive diffusion but increases plasma protein binding, creating competing effects on tissue penetration and volume of distribution.
Methodological Approach: Buccal Permeability Assay
In Vitro Buccal Permeability Protocol (Adapted from Patel et al.) [27]:
Tissue Preparation:
- Obtain fresh porcine buccal tissue immediately after sacrifice
- Separate buccal epithelium from underlying connective tissue
- Trim to standardized thickness (500 ± 50 μm)
- Maintain in phosphate buffer (pH 7.4) during transport and processing
- Initiate permeation studies within 2 hours of tissue isolation
Permeation Study Setup:
- Use horizontal, water-jacketed side-by-side diffusion cells
- Maintain temperature at 37°C with jacketed water circulation
- Set diffusional area to 0.68 cm²
- Mount tissue between donor and receiver chambers
- Equilibrate with phosphate buffer (pH 6.8 donor, pH 7.4 receiver) for 30 minutes
Experimental Conditions:
- Replace donor contents with drug solution in phosphate buffer (pH 6.8)
- Use saturated solutions for poorly soluble drugs
- For soluble drugs, use standardized concentration (e.g., 1.0-10 mg/ml)
- Stir both chambers with magnetic stir bars to minimize unstirred water layers
- Conduct experiments in triplicate
Sample Collection and Analysis:
- Collect receiver chamber samples at predetermined intervals over 5-8 hours
- Analyze drug content using validated HPLC methods with UV detection
- Use appropriate mobile phases and C18 columns for separation
- Calculate steady-state flux (Jss) from linear portion of cumulative amount versus time plot
Permeability Calculation:
- Apply equation: Kp = Jss / Cdonor
- Where Kp = apparent permeability coefficient (cm/s)
- Jss = steady-state flux (μg·h⁻¹·cm⁻²)
- Cdonor = initial donor concentration (μg/ml)
Volume of Distribution
Theoretical Framework
Volume of distribution (Vd) is a pharmacokinetic parameter that relates the total amount of drug in the body to its plasma concentration [32]. It represents the apparent volume into which a drug distributes to produce the observed plasma concentration [32]. Mathematically, Vd = Amount of drug in body / Plasma drug concentration [32] [30].
Vd is a theoretical concept that does not correspond directly to a physiological volume, but rather reflects the extent of tissue distribution relative to plasma concentration [32]. Drugs with high Vd values indicate extensive tissue distribution, while low Vd suggests confinement primarily to the plasma compartment [32] [30].
Lipophilicity-Vd Relationship
Lipophilicity directly influences volume of distribution by enhancing tissue penetration and binding [32] [25]. Lipophilic drugs typically display larger Vd values due to their ability to cross membranes and distribute into lipid-rich tissues [30]. However, extremely high lipophilicity may paradoxically reduce effective distribution due to extensive plasma protein binding that limits the free fraction available for tissue uptake [28].
The relationship follows a generally positive correlation, with Vd increasing as logD rises, though the slope varies significantly between drug classes and is modulated by factors such as ionization state, active transport processes, and tissue-specific binding [32]. In critically ill patients or those with pathophysiological changes, these relationships may be altered due to factors like fluid shifts, pH changes, and altered protein levels [26] [32].
Table 3: Volume of Distribution Classification and Lipophilicity Influence
| Vd Classification | Vd Range (L/kg) | Typical Lipophilicity | Distribution Pattern | Clinical Examples |
|---|---|---|---|---|
| Small | <0.25 | Low (logD <1) | Primarily plasma compartment | Gentamicin, Heparin [30] |
| Moderate | 0.25-1.0 | Low to moderate | Extracellular fluid | Theophylline, Aminophylline |
| Large | 1.0-5.0 | Moderate to high | Total body water | Digoxin, Amikacin |
| Very Large | >5.0 | High to very high | Extensive tissue sequestration | Chloroquine, Amitriptyline [27] [30] |
Methodological Approach: Determining Volume of Distribution
Experimental Protocol for Vd Determination (Adapted from Derangedphysiology.com) [32]:
Study Design:
- Administer precise intravenous dose to avoid absorption confounders
- Use radiolabeled compound or specific analytical method for accurate quantification
- Collect serial blood samples at predetermined timepoints
- Ensure adequate sampling duration to characterize distribution and elimination phases
Non-Compartmental Analysis (Varea):
- Measure area under the concentration-time curve (AUC) from time zero to infinity
- Determine terminal elimination rate constant (β) from slope of log-linear phase
- Apply formula: Varea = Dose / (AUC × β)
Compartmental Analysis (Vss):
- Employ multi-compartment modeling to account for distribution kinetics
- Calculate Vss using statistical moment theory: Vss = Dose × AUMC / AUC²
- Where AUMC = area under the first moment curve
Specialized Techniques:
- Use tissue homogenization and drug quantification for specific organ distribution
- Apply whole-body autoradiography for spatial distribution mapping
- Employ microdialysis for free drug concentration measurement in tissues
Data Interpretation:
- Relate Vd values to physiological volumes for context
- Consider impact of protein binding on interpretation
- Evaluate relationship between Vd and elimination half-life
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Materials for Distribution Studies
| Reagent/Material | Specifications | Research Application | Key Considerations |
|---|---|---|---|
| Porcine buccal tissue | Fresh, 500 ± 50 μm thickness | Permeability studies [27] | Requires immediate processing; viable within 2 hours post-isolation |
| Side-by-side diffusion cells | Water-jacketed, 0.68 cm² diffusional area | In vitro permeability assays [27] | Maintain 37°C; magnetic stirring to minimize unstirred water layers |
| Human serum albumin | Pharmaceutical grade, 35-40 g/L concentration | Plasma protein binding studies [28] | Represents primary binding protein for acidic drugs |
| α₁-acid glycoprotein | Purified, 0.4-1.0 g/L concentration | Plasma protein binding studies [28] | Critical for basic drug binding; acute phase reactant |
| C18 chromatography columns | 4.6 × 150 mm, 5 μm particle size | HPLC analysis of drug concentrations [27] | Enables precise drug quantification in permeability/protein binding assays |
| Phosphate buffers | pH 6.8 and 7.4, isotonic | Physiological simulation in assays [27] | pH 6.8 mimics oral cavity; pH 7.4 mimics plasma |
| n-octanol and buffer systems | HPLC grade solvents | Partition coefficient measurements [27] [5] | Standardized system for lipophilicity determination |
| Equilibrium dialysis devices | Molecular weight cutoff 12-14 kDa | Plasma protein binding assays [28] | Allows separation of free and bound drug fractions |
Integrated Experimental Workflow
Figure 2: Integrated workflow for evaluating distribution properties begins with lipophilicity assessment, proceeds through parallel permeability and protein binding assays, and culminates in volume of distribution determination to guide compound optimization.
The influence of lipophilicity on drug distribution represents a critical consideration in pharmaceutical research and development. Through its interconnected effects on plasma protein binding, tissue penetration, and volume of distribution, lipophilicity serves as a master variable governing a compound's pharmacokinetic profile. The relationships documented in this technical guide demonstrate that optimal distribution properties typically occur within a moderate lipophilicity range (logD ~1-3), balancing adequate membrane permeability with acceptable free fraction availability. For researchers engaged in drug design, systematic evaluation of these parameters using the methodologies outlined provides a rational framework for compound optimization. Future advances in this field will likely focus on refined predictive models that incorporate additional molecular descriptors and physiological considerations to further enhance our ability to design compounds with optimal distribution characteristics.
The blood-brain barrier (BBB) represents one of the most significant challenges in central nervous system (CNS) drug development, excluding over 98% of small-molecule drugs and all macromolecular therapeutics from accessing the brain. This technical review examines the crucial role of lipophilicity as a fundamental physicochemical property governing drug permeation across the BBB. Within the broader context of ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) research, we analyze how lipophilicity impacts critical pharmacokinetic parameters while balancing therapeutic efficacy with potential toxicity. The review integrates current experimental methodologies for lipophilicity assessment, advanced brain-targeting strategies that transcend traditional lipophilicity optimization, and computational approaches for predicting BBB permeability. As CNS drug discovery evolves toward multifunctional nanocarriers and targeted delivery systems, understanding and optimizing lipophilicity remains paramount for developing effective neurological therapeutics.
The blood-brain barrier is a highly selective semi-permeable membrane that separates circulating blood from the brain extracellular fluid, maintaining the delicate homeostasis required for proper CNS function. This protective interface consists of specialized endothelial cells connected by tight junctions, surrounded by pericytes, astrocytes, and basal lamina that collectively form a sophisticated neurovascular unit [33] [34]. The BBB's physiological role in protecting the brain from toxins and pathogens becomes a formidable obstacle for drug delivery, as it restricts the passage of most therapeutic agents [34].
From a drug development perspective, the BBB presents two primary challenges: limited passive diffusion due to tight junctions that restrict paracellular transport, and active efflux mechanisms that pump compounds back into the bloodstream [33] [35]. The tight junctions between cerebral endothelial cells significantly reduce paracellular permeability, while specialized transport systems and efflux transporters actively control transcellular passage [34]. These protective mechanisms explain why only small (<400-600 Da), lipophilic molecules with minimal hydrogen bonding capacity can passively diffuse across the BBB in therapeutically relevant concentrations [34].
Within ADMET research, lipophilicity emerges as a critical determinant of a compound's ability to navigate these barriers. As a key physicochemical parameter, lipophilicity influences virtually all pharmacokinetic processes, from initial absorption and distribution to metabolism, elimination, and potential toxicity [36] [11] [6]. The optimal balancing of lipophilicity represents one of the fundamental challenges in CNS drug design, requiring careful navigation between sufficient BBB permeability and acceptable safety profiles.
Lipophilicity Fundamentals and ADMET Relationships
Defining Lipophilicity Parameters
Lipophilicity, most commonly quantified as the partition coefficient (Log P) or distribution coefficient (Log D), describes a compound's equilibrium distribution between aqueous and lipid phases [37]. Log P represents the logarithm of the ratio of a compound's concentration in a neutral form between octanol and water, providing a measure of intrinsic lipophilicity independent of ionization. In contrast, Log D accounts for ionization at specific pH values (typically pH 7.4 for physiological relevance), making it more representative of distribution under biological conditions [11] [37]. These parameters serve as critical predictors of membrane permeability, with the gold standard measurement being the shake-flask method using octanol/water partitioning systems [11].
The relationship between lipophilicity and BBB permeability follows a well-established pattern, where passive diffusion is favored for compounds with moderate lipophilicity. However, this relationship is not linear, as excessive lipophilicity can diminish CNS exposure through increased plasma protein binding, heightened metabolism, and reduced aqueous solubility [36]. The optimal lipophilicity range for CNS drugs typically falls between Log P of 1-4, balancing sufficient membrane permeability with acceptable solubility and metabolic stability [36] [37].
Lipophilicity in the ADMET Framework
The influence of lipophilicity extends throughout the entire ADMET spectrum, making it a pivotal consideration in CNS drug design. The following table summarizes key ADMET relationships with lipophilicity:
Table 1: Lipophilicity-ADMET Relationships in CNS Drug Development
| ADMET Parameter | Relationship with Lipophilicity | Optimal Range (Log P) | Clinical Implications |
|---|---|---|---|
| Absorption | Increases with moderate lipophilicity due to enhanced membrane permeability | 1-3 | Facilitates gastrointestinal absorption and BBB crossing |
| Distribution | Higher lipophilicity increases volume of distribution and tissue binding | 2-4 | Enhances brain penetration but may increase off-target tissue accumulation |
| Metabolism | Elevated lipophilicity accelerates oxidative metabolism | <5 | Reduces half-life; increases risk of toxic metabolite formation |
| Excretion | Highly lipophilic compounds exhibit slower renal clearance | >0 | Prolonged systemic exposure potential; increased hepatobiliary elimination |
| Toxicity | Excessive lipophilicity correlates with promiscuous binding and cytotoxicity | <5 | Reduces therapeutic index; increases risk of phospholipidosis and organ toxicity |
The impact of lipophilicity on distribution processes is particularly relevant for CNS targeting. While moderate lipophilicity enhances passive diffusion across the BBB, excessive lipophilicity (Log P > 5) often leads to increased plasma protein binding and rapid hepatic metabolism, effectively reducing the fraction of freely available drug capable of crossing the BBB [36] [11]. Furthermore, highly lipophilic compounds demonstrate greater affinity for lipid membranes and increased risk of phospholipidosis, potentially contributing to cellular toxicity [36] [6].
The critical balance lies in achieving sufficient lipophilicity for BBB penetration while maintaining adequate aqueous solubility for formulation and dissolution. This balance is quantified through lipophilic efficiency metrics, such as LipE (Lipophilic Efficiency), which normalize potency against lipophilicity to guide compound optimization [36]. Monitoring these indices throughout drug discovery helps maintain optimal physicochemical properties associated with successful CNS therapeutics.
Experimental Methodologies for Lipophilicity Assessment
Conventional Measurement Techniques
Accurate determination of lipophilicity remains essential for CNS drug development, with several established methodologies providing experimental data. The shake-flask method, recognized as the gold standard, involves direct measurement of compound distribution between octanol and aqueous buffer phases under controlled conditions [11]. While highly accurate, this approach is time-consuming, requires relatively pure compounds in substantial quantities, and is limited to a Log P range of approximately -2 to 4 [11].
Chromatographic techniques offer efficient alternatives for lipophilicity assessment. Reversed-phase thin-layer chromatography (RP-TLC) and reversed-phase high-performance liquid chromatography (RP-HPLC) provide indirect measurements through correlation of retention factors with lipophilicity parameters [11]. These methods require minimal compound quantities, offer high throughput capacity, and demonstrate good reproducibility, with accuracy typically within ±1 unit compared to shake-flask values [11]. The chromatographically-derived lipophilicity parameter (Rₘ⁰) serves as a reliable experimental descriptor for quantitative structure-activity relationship studies.
Advanced and High-Throughput Approaches
Contemporary drug discovery employs immobilized artificial membrane (IAM) chromatography and immobilized liposome chromatography (ILC) to better mimic biological membrane interactions [37]. These techniques provide chromatographic indices that correlate with drug partitioning into lipid bilayers, potentially offering more biologically relevant permeability predictions than octanol/water systems.
The experimental workflow for comprehensive lipophilicity assessment typically follows a tiered approach:
Diagram 1: Experimental lipophilicity assessment workflow.
For ionizable compounds, pH-dependent distribution coefficients (Log D) provide more physiologically relevant data than partition coefficients (Log P). Determining Log D at pH 7.4 (blood pH) and 6.5 (intestinal pH) offers insights into behavior under different biological environments [37]. However, reliable computational prediction of Log D remains challenging, emphasizing the importance of experimental measurement for critical compounds [37].
Beyond Simple Lipophilicity: Advanced CNS Targeting Strategies
Limitations of Lipophilicity-Optimized Compounds
While moderate lipophilicity remains necessary for passive BBB penetration, it is insufficient for many CNS therapeutics. Several limitations emerge with reliance solely on lipophilicity optimization. Numerous lipophilic compounds act as substrates for efflux transporters, particularly P-glycoprotein (P-gp), which actively pumps drugs back into the bloodstream, substantially reducing brain exposure [35] [38]. This efflux mechanism explains why many compounds with favorable lipophilicity demonstrate poor CNS penetration in vivo [38].
Excessive lipophilicity also correlates with increased metabolic clearance, poor aqueous solubility, and non-specific tissue binding, potentially leading to higher toxicity and limited therapeutic utility [36]. Furthermore, many modern therapeutic modalities (peptides, proteins, nucleic acids) cannot be optimized for BBB penetration through simple lipophilicity adjustments, necessitating alternative delivery strategies.
Novel Brain-Targeted Delivery Platforms
Advanced drug delivery systems have emerged to overcome BBB limitations while mitigating lipophilicity-related challenges. Nanoparticle-based systems, including liposomes, polymeric nanoparticles, and solid lipid nanoparticles, can encapsulate both hydrophilic and lipophilic compounds, protecting them from metabolism and efflux while facilitating brain delivery [33] [39]. These nanocarriers can be surface-functionalized with targeting ligands to exploit endogenous transport pathways.
Receptor-mediated transcytosis represents a particularly promising approach for biologics delivery. By conjugating therapeutics or nanocarriers to ligands targeting receptors highly expressed on BBB endothelial cells (transferrin receptor, insulin receptor, low-density lipoprotein receptor), drugs can hijack natural transport mechanisms for CNS delivery [39] [40]. Transferrin receptor-targeted nanoparticles, for example, have demonstrated enhanced brain uptake of therapeutic agents in preclinical models of Alzheimer's disease and glioblastoma [39].
Additional innovative strategies include:
- Cell-mediated transcytosis: Utilizing monocytes or macrophages as carrier cells for CNS delivery
- Focus ultrasound with microbubbles: Temporarily disrupting BBB integrity to enhance local drug penetration
- Intranasal administration: Bypassing the BBB entirely via olfactory and trigeminal neural pathways
- Prodrug approaches: Designing lipophilic precursors that convert to active drugs after BBB penetration
The following diagram illustrates multifactorial BBB penetration strategies:
Diagram 2: Multifactorial BBB penetration strategies.
Computational Prediction and Optimization Frameworks
In Silico Lipophilicity and BBB Permeability Models
Computational approaches have become indispensable for predicting lipophilicity and BBB permeability during early drug discovery. Multiple algorithms exist for calculating partition coefficients, including fragment-based methods (ClogP, ACD/LogP), atom-based approaches (AlogP, XLOGP), and property-based methods (MLOGP) [11] [6]. These tools enable rapid virtual screening of compound libraries, prioritizing candidates with optimal lipophilicity ranges before synthesis.
Machine learning models have demonstrated particular utility in predicting BBB permeability by integrating multiple physicochemical descriptors beyond lipophilicity. These models typically incorporate parameters such as molecular weight, polar surface area, hydrogen bond donors/acceptors, rotatable bonds, and pKa values to generate more accurate predictions [39] [6]. The SwissADME and pkCSM web tools provide comprehensive profiling of ADMET parameters, including BBB penetration predictions, enabling researchers to identify potential development challenges early [11].
Recent advancements integrate artificial intelligence with molecular modeling to optimize brain-targeted chemical entities. These systems can propose structural modifications that balance lipophilicity with other critical parameters, suggesting specific functional group changes to enhance BBB penetration while maintaining target engagement [39] [40].
Integrated Property-Based Design Rules
Successful CNS drugs generally follow established physicochemical property guidelines that extend beyond lipophilicity alone. These include:
- Molecular weight < 450 Da
- ClogP between 1-4
- Polar surface area < 90 Ų
- Hydrogen bond donors ≤ 3
- Hydrogen bond acceptors ≤ 7
- Rotatable bonds ≤ 10
While these rules provide valuable guidance, they represent generalizations rather than absolute constraints. Property-based design must consider the specific therapeutic target, mechanism of action, and intended dosing regimen. The emerging paradigm emphasizes maintaining compounds within a defined "property space" associated with successful CNS drugs rather than strict adherence to individual parameter thresholds [36] [6].
Research Reagent Solutions for CNS Targeting Studies
Table 2: Essential Research Tools for BBB Permeability and Lipophilicity Assessment
| Category | Specific Reagents/Tools | Research Application | Key Features |
|---|---|---|---|
| Lipophilicity Measurement | Octanol-buffer systems; RP-18 TLC/HPLC columns; IAM chromatography columns | Experimental determination of Log P/Log D | Gold-standard partitioning; High-throughput screening; Biomimetic membrane partitioning |
| Computational Prediction | SwissADME; pkCSM; Chemaxon; Molinspiration | In silico property prediction | Multi-parameter ADMET profiling; Log P calculation; BBB permeability prediction |
| BBB Permeability Models | Parallel artificial membrane permeability assay (PAMPA-BBB); MDCK-MDR1 cell lines; In vitro BBB co-culture models | Permeability screening | High-throughput passive permeability; Active transport and efflux assessment; Physiological BBB mimicry |
| Brain-Targeting Nanocarriers | PLGA nanoparticles; Liposomes with surface functionalization; Solid lipid nanoparticles | Advanced delivery system evaluation | Drug encapsulation and release; Surface modification for targeting; Controlled biodistribution |
| Targeting Ligands | Transferrin; Lactoferrin; Anti-TfR antibodies; Cell-penetrating peptides | Receptor-mediated transcytosis studies | BBB receptor targeting; Enhanced cellular uptake; Improved brain distribution |
Lipophilicity remains a cornerstone parameter in CNS drug design, fundamentally influencing a compound's ability to navigate the blood-brain barrier while simultaneously impacting broader ADMET properties. The optimal navigation of lipophilicity-space requires balancing passive permeability with solubility, metabolic stability, and efflux transporter susceptibility. While traditional approaches focused primarily on optimizing Log P, contemporary strategies increasingly leverage advanced delivery systems that circumvent BBB limitations through targeted transport mechanisms.
The future of CNS-targeted therapeutics lies in integrated approaches that combine thoughtful physicochemical property optimization with sophisticated delivery technologies. Multifunctional nanocarriers with targeting ligands, stimuli-responsive release mechanisms, and efflux pump inhibition capabilities represent the next frontier in brain drug delivery. Furthermore, the continued refinement of computational models and AI-driven design tools will accelerate the identification of CNS-accessible chemical space, potentially expanding the therapeutic landscape for neurological disorders. As our understanding of BBB biology and drug transport mechanisms evolves, so too will our ability to precisely engineer therapeutics that successfully navigate this formidable barrier.
Correlations with Metabolic Stability and Cytochrome P450 Enzyme Interactions
Metabolic stability, a cornerstone of pharmacokinetics, dictates the in vivo half-life and bioavailability of drug candidates. A primary determinant of this stability is their interaction with the Cytochrome P450 (CYP) enzyme system, the body's most important machinery for Phase I drug metabolism. This whitepaper explores the intrinsic correlation between metabolic stability and CYP-mediated metabolism, framing this relationship within the critical context of lipophilicity and its overarching influence on ADMET properties. We provide a detailed examination of the molecular mechanisms of enzyme inhibition and induction, the impact of genetic polymorphisms, and the advanced in silico and in vitro methodologies employed in contemporary drug discovery to optimize these parameters, thereby reducing late-stage attrition due to pharmacokinetic failures.
The Cytochrome P450 (CYP) system represents a superfamily of heme-containing enzymes predominantly located in the liver, though they are also present in the gastrointestinal tract, lungs, and kidneys, contributing to extrahepatic metabolism [41] [42]. These enzymes are membrane-bound proteins located in the smooth endoplasmic reticulum and are essential for the oxidative biotransformation of a vast array of exogenous and endogenous compounds [41]. Of the more than 50 identified human CYP enzymes, six are responsible for metabolizing approximately 90% of all clinically relevant drugs: CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 [41] [42]. Of these, CYP3A4 is the most abundant and is involved in the metabolism of nearly half of all modern pharmaceuticals [41].
Metabolic stability refers to the resistance of a compound to enzymatic degradation. A drug's metabolic stability is a key predictor of its intrinsic clearance and in vivo half-life [43]. Compounds with low metabolic stability are rapidly cleared, often leading to insufficient exposure and therapeutic failure. Conversely, excessive stability can lead to accumulation and potential toxicity. The metabolism of drugs via the cytochrome P450 system has emerged as an important determinant in the occurrence of several drug interactions that can result in drug toxicities, reduced pharmacological effect, and adverse drug reactions [41]. Therefore, understanding and optimizing a compound's interaction with CYP enzymes is a fundamental objective in early-stage drug discovery.
The Lipophilicity-ADMET Nexus and CYP Interactions
Lipophilicity, typically quantified as the partition coefficient (LogP), is one of the principal parameters describing a drug's pharmacokinetic behavior, including its absorption, distribution, metabolism, excretion, and toxicity (ADMET) [15]. Medicinal substances with moderate lipophilicity tend to be better absorbed through cell membranes, which can affect their rate and absorption efficiency from the gastrointestinal tract [15].
However, the relationship between lipophilicity and metabolic stability is complex. While increased lipophilicity can enhance membrane permeability, it also often renders a molecule more susceptible to metabolism in the liver through oxidation, reduction, and conjugation reactions [15]. Highly lipophilic compounds are more likely to be substrates for CYP-mediated oxidation, as these enzymes have evolved to process hydrophobic xenobiotics. The impact of lipophilicity on metabolism can have direct consequences for pharmacological activity and toxicity [15]. For instance, lipophilicity influences a drug's distribution and accumulation in lipid-rich tissues, which can affect its availability to hepatic CYP enzymes and its overall excretion profile [15]. Consequently, during lead optimization, medicinal chemists often aim to fine-tune lipophilicity to strike a balance between favorable permeability and acceptable metabolic stability, thereby reducing the risk of drug-drug interactions and toxicity.
Molecular Mechanisms of CYP Enzyme Interactions
Drugs can interact with the CYP system in three primary ways: as substrates, inhibitors, or inducers. The interplay between these roles is a common source of clinically significant drug-drug interactions.
- Enzyme Substrates: A substrate is a drug that is metabolized by a specific CYP enzyme. Some drugs are metabolized by only one CYP enzyme (e.g., metoprolol by CYP2D6), while others are metabolized by multiple enzymes (e.g., warfarin by CYP1A2, CYP2D6, and CYP3A4) [42]. This distinction is critical; a drug that is a substrate for a single enzyme is more vulnerable to interactions with inhibitors of that pathway.
- Enzyme Inhibition: Inhibition occurs when one drug blocks the metabolic activity of a CYP enzyme, leading to increased serum levels of a second drug that is a substrate for the same enzyme [41]. For most drugs, this can lead to a greater potential for toxicity [41]. Inhibition can be competitive (both drugs competing for the same enzyme active site) or mechanism-based (inactivation of the enzyme). Inhibitory effects usually occur immediately upon administration of the inhibitor [42].
- Enzyme Induction: Induction is the process by which a drug increases the synthesis and activity of CYP enzymes, resulting in accelerated biotransformation and reduced plasma concentrations of co-administered drugs [41]. Unlike inhibition, the onset of induction is delayed, depending on the half-life of the inducing drug, as it requires new enzyme synthesis [42]. This can lead to therapeutic failure.
The following table summarizes key clinical examples of substrates, inhibitors, and inducers for the major CYP enzymes:
Table 1: Key CYP Enzymes and Their Interacting Drugs
| CYP Enzyme | Select Substrates | Potent Inhibitors | Potent Inducers |
|---|---|---|---|
| CYP3A4 | Alprazolam, cyclosporine, simvastatin [42] [44] | Clarithromycin, itraconazole, ritonavir [42] [44] | Carbamazepine, phenytoin, rifampin [42] |
| CYP2D6 | Codeine, metoprolol, paroxetine [42] | Fluoxetine, paroxetine, quinidine [42] | No significant inducers [42] |
| CYP2C9 | Celecoxib, glipizide, warfarin [42] [44] | Amiodarone, fluconazole, fluoxetine [42] | Carbamazepine, rifampin [42] |
| CYP2C19 | Omeprazole, phenytoin [42] | Fluvoxamine, isoniazid, ritonavir [42] | Carbamazepine, rifampin [42] |
| CYP1A2 | Caffeine, clozapine, theophylline [42] | Ciprofloxacin, fluvoxamine [42] | Carbamazepine, tobacco [42] |
Experimental Protocols for Assessing Metabolic Stability and CYP Interactions
Rigorous in vitro screening is standard practice in Discovery DMPK (Drug Metabolism and Pharmacokinetics) to identify potential metabolic liabilities early in the development process.
In Vitro Metabolic Stability Assays
These assays are designed to determine a compound's intrinsic clearance (CL~int~) and are typically automated for medium- to high-throughput screening [43].
Protocol Summary:
- Incubation Setup: The test compound (at a concentration typically ≤1-3 μM) is incubated with a metabolically active system, such as human liver microsomes or cryopreserved hepatocytes (e.g., 0.25–1 mg microsomal protein/ml or 0.25–1 × 10^6 cells/ml) [43].
- Reaction Conditions: The incubation is carried out in a suitable buffer (e.g., phosphate or TRIS) supplemented with nicotinamide adenine dinucleotide phosphate (NADPH) to provide the necessary cofactors for oxidative metabolism. Solvent concentrations (e.g., acetonitrile or DMSO) should be minimized (≤1%, v/v) to avoid inhibiting CYP activity [43].
- Sampling: Aliquots of the incubation mixture are taken at predetermined time points (e.g., 0, 5, 15, 30, 45, 60 minutes).
- Termination and Analysis: Reactions are terminated by adding an organic solvent like acetonitrile. The samples are centrifuged to precipitate proteins, and the supernatant is analyzed using techniques like LC-MS/MS to determine the parent compound's concentration over time [43].
- Data Analysis: The half-life (t~1/2~) and intrinsic clearance (CL~int~) are calculated from the exponential decay of the parent compound.
CYP Inhibition Screening
This frontline screen assesses a new compound's potential to cause drug-drug interactions by inhibiting key CYP enzymes.
Protocol Summary:
- Enzyme Source: Use human liver microsomes or recombinant CYP enzymes.
- Probe Reactions: Incubate the enzyme source with a well-established substrate for a specific CYP isoform (e.g., phenacetin for CYP1A2, dextromethorphan for CYP2D6, midazolam for CYP3A4) in the presence and absence of the test compound.
- Inhibitor Concentration: The test compound is evaluated at a range of concentrations to determine the IC~50~ value (concentration that inhibits 50% of enzyme activity).
- Metabolite Quantification: The formation of the specific metabolite from the probe substrate is measured, typically using LC-MS/MS. A decrease in metabolite formation in the presence of the test compound indicates inhibition.
- Data Analysis: IC~50~ values are determined, and compounds are classified as strong, moderate, or weak inhibitors based on regulatory guidelines.
Table 2: Essential Research Reagents for In Vitro DMPK Studies
| Reagent / Tool | Function in Experimentation |
|---|---|
| Human Liver Microsomes | A subcellular fraction containing membrane-bound CYP enzymes; used for metabolic stability and CYP inhibition assays [43]. |
| Cryopreserved Hepatocytes | Intact human liver cells that contain the full complement of drug-metabolizing enzymes and cofactors; used for more physiologically relevant clearance and induction studies [43]. |
| Recombinant CYP Enzymes | Individual CYP isoforms expressed in cell systems; used for reaction phenotyping to identify which specific enzyme metabolizes a drug [43]. |
| LC-MS/MS (Liquid Chromatography-Tandem Mass Spectrometry) | The primary analytical platform for quantifying drugs and their metabolites in complex biological matrices due to its high sensitivity and specificity [43]. |
| Specific Probe Substrates | Well-characterized drugs metabolized primarily by a single CYP enzyme (e.g., midazolam for CYP3A4); used to monitor the activity of that specific enzyme in inhibition assays [43]. |
Advanced In Silico and Federated Learning Approaches
The advent of sophisticated computational models has dramatically enhanced the predictive power of ADMET profiling.
- In Silico ADMET Prediction Platforms: Tools like SwissADME, pkCSM, and ADMETlab 2.0 are widely used to predict physicochemical properties, drug-likeness, and pharmacokinetic parameters, including CYP inhibition profiles [15] [45] [46]. These platforms use various algorithms (e.g., iLOGP, XLOGP3) to estimate lipophilicity (LogP) and other critical descriptors, allowing for rapid virtual screening of large compound libraries during the early stages of drug development [15].
- Federated Learning for ADMET Prediction: A major limitation of isolated modeling efforts is that each organization's data captures only a small fraction of chemical space. Federated learning overcomes this by enabling collaborative model training across distributed, proprietary datasets without centralizing the data, thus preserving privacy and intellectual property [47]. This approach systematically extends the model's effective domain, leading to higher accuracy and broader applicability, particularly for pharmacokinetic and safety endpoints [47]. Multi-task architectures trained on such diverse data have achieved up to 40–60% reductions in prediction error for endpoints like metabolic clearance [47].
- Machine Learning Models: Advanced deep learning architectures, such as Transformer-based models (e.g., MSformer-ADMET), are now being specialized for ADMET prediction. These models can effectively model long-range dependencies in molecular representations and have been shown to outperform conventional methods across a wide range of ADMET endpoints [48].
Visualization of Workflows and Relationships
The following diagram illustrates the interconnected relationship between lipophilicity, CYP-mediated metabolism, and ADMET outcomes, as well as the key experimental workflow for evaluation.
Diagram 1: Lipophilicity, CYP interactions, and metabolic stability relationships with experimental workflow.
The correlation between metabolic stability and Cytochrome P450 enzyme interactions is a fundamental aspect of drug design that cannot be disentangled from the critical influence of lipophilicity on the overall ADMET profile. A comprehensive understanding of whether a drug candidate acts as a CYP enzyme substrate, inducer, or inhibitor is paramount to predicting and preventing clinically significant drug interactions and toxicities. The integrated use of robust in vitro screening protocols, cutting-edge in silico prediction tools, and collaborative modeling approaches like federated learning provides a powerful framework for optimizing these properties early in the drug discovery pipeline. By systematically applying these principles and technologies, researchers can more efficiently advance drug candidates with a higher probability of clinical success, ultimately reducing the high attrition rates attributed to unfavorable pharmacokinetics and safety profiles.
Lipophilicity, most commonly measured as the logarithmic n-octanol-water partition coefficient (logP), represents one of the most fundamental physicochemical properties in drug design and development. This parameter quantitatively expresses a compound's solubility in non-polar solvents versus water and consequently determines its ability to passively penetrate biological membranes [49] [5]. The critical importance of lipophilicity extends throughout the entire ADMET spectrum—affecting Absorption, Distribution, Metabolism, Excretion, and Toxicity profiles of potential drug candidates. Research has consistently demonstrated that optimal lipophilicity values are essential for balancing permeability and solubility, with excessively high logP values frequently correlating with poor aqueous solubility, increased metabolic turnover, tissue accumulation, and heightened risks of specific toxicity mechanisms [5] [50].
Within the broader context of ADMET research, two particularly problematic toxicity risks associated with elevated lipophilicity are hERG channel inhibition and drug-induced phospholipidosis (DIPL). The human Ether-à-go-go Related Gene (hERG) potassium channel plays a critical role in cardiac repolarization, and its inhibition by small molecules can prolong the QT interval, potentially leading to fatal arrhythmias like Torsades de Pointes [51] [52]. Similarly, DIPL is an acquired lysosomal storage disorder characterized by excessive accumulation of phospholipids, which can lead to organ toxicity in heart, liver, lungs, and kidneys [53] [54]. This technical guide examines the mechanistic connections between high lipophilicity and these specific toxicity risks, providing researchers with experimental protocols, computational approaches, and mitigation strategies essential for modern drug development.
Quantitative Relationships: Lipophilicity and Toxicity Risks
Structural and Physicochemical Correlations with Toxicity
Computational analyses across diverse compound datasets reveal distinct trends in physicochemical properties for molecules associated with specific toxicity risks. Protein-protein interaction inhibitors (iPPIs), which often face development challenges due to toxicity concerns, exhibit the highest mean molecular weight (521 Da) and elevated mean logP values (4.8) compared to other compound classes [50]. This combination of high molecular weight and lipophilicity creates particular challenges for maintaining favorable ADMET profiles.
Table 1: Computed Physicochemical Properties Across Compound Classes
| Dataset | Mean MW (Da) | Mean logP | Mean HBD | Mean HBA | Mean TPSA (Ų) | Rotatable Bonds |
|---|---|---|---|---|---|---|
| iPPIs | 521 | 4.8 | 2.1 | 7.0 | 101 | 7 |
| Enzyme Inhibitors | 438 | 3.3 | 1.9 | 7.0 | 108 | 6 |
| GPCR Ligands | 427 | 3.9 | 1.8 | 5.6 | 77 | 6 |
| Ion Channel Modulators | 414 | 3.3 | 1.7 | 5.7 | 80 | 6 |
| Nuclear Receptor Ligands | 438 | 4.8 | 1.8 | 5.2 | 81 | 6 |
| Oral Marketed Drugs | 376 | 2.5 | 1.7 | 5.0 | 75 | 5 |
| Natural Product-Derived Drugs | 383 | 3.3 | 2.8 | 6.1 | 109 | 5 |
The correlation between lipophilicity and toxicity manifestations is particularly evident in comparative analyses. Compounds with high logP values (>5) demonstrate increased propensity for hERG channel inhibition and phospholipidosis, with nuclear receptor ligands and iPPIs showing the highest mean logP values at 4.8 [50]. This trend follows for logD values (logP corrected for pKa of ionizable groups) with the highest values for nuclear receptor compounds and iPPIs (mean logD 3.8 and 3.5, respectively) [50].
Experimental Lipophilicity Measurements
Reverse-phase thin-layer chromatography (RP-TLC) provides valuable experimental lipophilicity measurements (RM0) that complement computational predictions. In studies of quinoline-1,4-quinone hybrids, experimental RM0 values ranged from 1.51 to 3.39, demonstrating the method's sensitivity to structural modifications [49]. The relationship between retention parameter (Rf) and lipophilicity follows the equation:
RM = log(1/Rf - 1)
where RM values are extrapolated to zero concentration of organic solvent to determine the chromatographic lipophilicity parameter (RM0) using the equation:
RM = RM0 + bC
(C represents the concentration of acetone in the mobile phase, while b is the slope of the regression plot) [49]. These experimental values provide crucial validation for computational models and establish more reliable structure-activity relationships.
hERG Channel Inhibition: Mechanisms and Detection
Molecular Mechanisms of hERG Inhibition
The hERG (Kv11.1) potassium channel plays a critical role in the repolarization phase of the cardiac action potential. Structurally diverse small molecules can block this channel by binding to its inner cavity, particularly to specific amino acid residues like F656 and Y652 [52]. This blockade reduces the outward potassium current during cardiac repolarization, leading to prolongation of the QT interval on electrocardiograms, which can progress to Torsades de Pointes and sudden cardiac death [51] [52]. The hERG channel's unusual structural features, including a large inner vestibule and specific aromatic residues, make it particularly susceptible to inhibition by promiscuous compounds with high lipophilicity.
Experimental and Computational Assessment
Traditional assessment of hERG liability has relied on a combination of in vitro and in vivo methods. In vitro methods include radioligand binding assays and patch clamp techniques, particularly the gold-standard electrophysiological patch clamp, which directly measures ion channel function but is costly and low-throughput [51]. In vivo hERG assays, while providing systemic physiological context, suffer from even lower throughput and higher costs, making them impractical for early-stage screening of large compound libraries [51].
Table 2: hERG Risk Assessment Methods
| Method Type | Specific Techniques | Key Outputs | Advantages | Limitations |
|---|---|---|---|---|
| In Vitro Assays | Patch clamp electrophysiology | IC50 values, current inhibition | Gold standard, direct functional measurement | Low throughput, high cost |
| Radioligand binding assays | Displacement constants | Higher throughput | Indirect measurement | |
| In Silico Prediction | XGBoost with ISE mapping | Classification (inhibitor/non-inhibitor) | High throughput, low cost | Model dependent |
| Deep learning neural networks | Probability scores | Handles complex patterns | Black box nature | |
| In Vivo Testing | QT interval prolongation in animals | ECG parameters | Physiological context | Very low throughput, ethical concerns |
Advanced computational approaches have emerged as valuable tools for early hERG risk assessment. Machine learning models, particularly eXtreme Gradient Boosting (XGBoost) integrated with Isometric Stratified Ensemble (ISE) mapping, have demonstrated competitive predictive performance with sensitivity of 0.83 and specificity of 0.90 [51]. These models utilize diverse molecular descriptors including peoe_VSA8, ESOL, SdssC, MaxssO, nRNR2, MATS1i, nRNHR, and nRNH2, which capture critical structural features associated with hERG inhibition [51]. The FDA's Comprehensive In Vitro Pro-Arrhythmia Assay (CiPA) initiative further promotes the integration of in silico models with in vitro data from engineered human cells and stem cell-derived cardiomyocytes for improved cardiotoxicity assessment in early drug development [51].
Figure 1: hERG Inhibition Toxicity Pathway. This diagram illustrates the sequential molecular and physiological events connecting high lipophilicity to potentially fatal cardiac outcomes through hERG channel inhibition.
Drug-Induced Phospholipidosis: Mechanisms and Detection
Molecular Mechanisms of Phospholipidosis
Drug-induced phospholipidosis (DIPL) is an acquired lysosomal storage disorder characterized by the excessive accumulation of phospholipids within lysosomes and the formation of characteristic lamellar bodies [53] [54]. The condition predominantly occurs with cationic amphiphilic drugs (CADs), which contain both a hydrophobic ring structure and a hydrophilic side chain with a cationic group [53] [55]. Over 200 marketed drugs are classified as CADs with potential to induce phospholipid deposition, including widely prescribed agents such as amiodarone, fluoxetine, gentamicin, hydroxychloroquine, perhexiline, and certain β-blockers like bisoprolol and carvedilol [53] [54].
The mechanistic basis of DIPL involves the lysosomotropic behavior of CADs. These compounds accumulate within acidic lysosomal compartments where they become protonated and trapped due to their weak basic properties—a process known as lysosomal trapping or acidotropy [55]. Once accumulated, CADs can inhibit lysosomal phospholipase activity or form drug-phospholipid complexes that resist degradation, ultimately leading to phospholipid accumulation and lamellar body formation [53] [54]. This disruption of lysosomal function can lead to cellular toxicity, with clinical manifestations depending on the affected tissues (heart, liver, lungs, kidneys).
Experimental and Computational Assessment
Multiple experimental approaches exist for detecting DIPL, ranging from traditional histopathology to modern high-content screening methods. Electron microscopy remains the gold standard for definitive identification of lamellar bodies in tissues, as demonstrated in a case report where post-mortem examination revealed vacuolar degeneration and lamellar body accumulation in the myocardium of a patient taking bisoprolol [53]. However, for early-stage screening, fluorescence-based assays using labeled phospholipids (e.g., LipidGreen2) combined with high-content live-cell imaging provide higher throughput capabilities [56].
Advanced machine learning approaches have been developed to predict PLD induction potential based on chemical structure. These models utilize comprehensive compound libraries and various algorithmic approaches, with interpretation techniques like SHapley Additive exPlanations (SHAP) providing insights into structural features contributing to PLD risk [56]. These computational tools are particularly valuable given that DIPL caused by drug combinations appears to occur in an approximately additive manner, emphasizing the importance of early risk identification in the context of polypharmacy [55].
Figure 2: Drug-Induced Phospholipidosis Pathway. This diagram illustrates the sequential molecular and cellular events through which cationic amphiphilic drugs induce phospholipid accumulation and subsequent organ toxicity.
Table 3: Research Reagent Solutions for Toxicity Assessment
| Tool Category | Specific Tools/Platforms | Application | Key Features |
|---|---|---|---|
| Computational Prediction | SwissADME | Lipophilicity and ADMET prediction | Multiple logP algorithms, bioavailability radar |
| pkCMS | ADMET parameter calculation | Comprehensive property profiling | |
| XGBoost with ISE mapping | hERG inhibition prediction | Handles class imbalance, confidence estimation | |
| KNIME with RDKit plugins | Workflow automation and descriptor calculation | Open-source, customizable pipelines | |
| Experimental Assays | RP-TLC | Experimental lipophilicity determination | Low-cost, high-throughput screening |
| Patch clamp electrophysiology | hERG channel function assessment | Gold standard, direct measurement | |
| High-content live-cell imaging | Phospholipidosis detection | Quantitative, high-throughput | |
| Electron microscopy | Lamellar body identification | Definitive structural confirmation | |
| Chemical Libraries | Chemogenomic libraries | Compound screening | Structurally diverse compounds |
| CAD databases | Phospholipidosis risk assessment | Known cationic amphiphilic drugs |
Experimental Protocols for Toxicity Assessment
RP-TLC Protocol for Experimental Lipophilicity Determination
The Reverse-Phase Thin Layer Chromatography (RP-TLC) method provides a reliable experimental approach for determining compound lipophilicity [49] [5]:
- Stationary Phase Preparation: Use modified silica gel (e.g., RP-18 or RP-8) as the stationary phase on TLC plates.
- Sample Application: Apply 5 μL of ethanolic compound solutions (1-24 test compounds and reference substances) to chromatographic plates using a micropipette.
- Mobile Phase Preparation: Prepare mixtures of tris(hydroxymethyl)aminomethane (TRIS) buffer (0.2 M, pH = 7.4) with acetone at varying concentrations (50%, 55%, 60%, 65%, 70%, 75%, and 80%).
- Chromatography Development: Develop chromatograms in appropriately saturated chambers using the prepared mobile phases.
- Visualization: Detect spots using iodine vapor or UV light for non-chromophoric compounds.
- Data Calculation: Measure retardation factors (Rf) and convert to RM values using the equation: RM = log(1/Rf - 1).
- Lipophilicity Determination: Extrapolate RM values to zero concentration of organic modifier to obtain the chromatographic lipophilicity parameter (RM0) using the equation: RM = RM0 + bC, where C is the concentration of organic solvent and b is the slope of the regression plot [49].
High-Content Live-Cell Imaging for Phospholipidosis Detection
A versatile high-content live-cell imaging approach provides robust quantification of phospholipidosis induction [56]:
- Cell Culture: Plate appropriate cell lines (e.g., HepG2 or primary macrophages) in multi-well plates at optimal density and culture until 70-80% confluent.
- Compound Treatment: Treat cells with test compounds at relevant concentrations (typically 1-10 μM) for 24-72 hours. Include positive controls (known PLD inducers like amiodarone) and negative controls.
- Staining: Incubate cells with fluorescent phospholipid probes (e.g., LipidGreen2) according to manufacturer protocols. Counterstain with lysosomal markers (e.g., LysoTracker) if performing co-localization studies.
- Image Acquisition: Acquire high-resolution images using automated high-content imaging systems with appropriate fluorescence channels.
- Image Analysis: Quantify fluorescence intensity and morphological changes using image analysis software. Machine learning algorithms can be trained to classify PLD-positive cells based on pattern recognition.
- Data Interpretation: Calculate fold-increase in phospholipid accumulation compared to negative controls. Establish threshold values for PLD classification based on positive controls and historical data.
The connection between high lipophilicity and toxicity risks represents a critical consideration in modern drug design. Through mechanistic understanding of hERG inhibition and phospholipidosis, coupled with robust experimental and computational assessment methods, researchers can effectively identify and mitigate these risks early in the drug development process. The integration of lipophilicity optimization strategies with comprehensive toxicity screening creates a powerful framework for advancing safer therapeutic candidates while minimizing late-stage attrition due to toxicity concerns. As predictive models continue to improve through machine learning and more comprehensive datasets, the field moves closer to in silico-first approaches for toxicity risk assessment, aligning with the 4R principles (reduce, refine, replace, responsibility) in pharmaceutical research and development.
Methodologies for Assessing and Applying Lipophilicity in Drug Discovery
Lipophilicity, the measure of a molecule's affinity for a lipid environment over an aqueous one, is a fundamental physicochemical property in drug discovery and development. It is most frequently expressed as the logarithm of the partition coefficient (log P) for neutral compounds or the distribution coefficient (log D) at a specific pH for ionizable species [14] [57]. This parameter is not merely a descriptor of solubility; it is a critical determinant of a compound's pharmacokinetic and pharmacodynamic behavior, directly influencing its absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile [58] [14] [57]. Poor lipophilicity-related characteristics are a leading cause of failure in drug development, contributing to inefficacy, toxicity, and escalating costs [14].
Within this context, the accurate determination of lipophilicity is paramount. Three experimental techniques have emerged as gold standards: the classical shake-flask method, reversed-phase thin-layer chromatography (RP-TLC), and reversed-phase high-performance liquid chromatography (RP-HPLC). This technical guide provides an in-depth examination of these core methodologies, detailing their principles, protocols, and applications in modern medicinal chemistry research focused on understanding and optimizing the ADMET properties of new chemical entities.
The Central Role of Lipophilicity in ADMET
Lipophilicity serves as a master variable that modulates every aspect of a drug's journey through the body. Its influence begins with absorption, as drugs must passively cross lipid biomembranes to reach their systemic circulation. The rate-limiting step for hydrophilic compounds (low log P) is partitioning into the hydrophobic membrane, while for hydrophobic compounds (high log P), it is partitioning back into the aqueous intracellular environment [57]. Furthermore, lipophilicity is inversely correlated with aqueous solubility, a key factor for bioavailability [57].
Regarding distribution, lipophilic compounds tend to have a larger volume of distribution and higher plasma protein binding, which can reduce the concentration of free, active drug [14] [57]. The ability to penetrate the blood-brain barrier (BBB) is also strongly linked to lipophilicity, with optimal log P values around 2 facilitating passive diffusion [14]. Beyond pharmacokinetics, lipophilicity influences pharmacodynamics by governing ligand-target interactions. It is a key parameter in Quantitative Structure-Activity Relationship (QSAR) studies [58] [14]. However, increasing lipophilicity to gain potency carries risks, including target promiscuity and a higher likelihood of toxicity, such as interaction with the hERG channel associated with cardiac arrhythmias [57]. Consequently, controlling lipophilicity within an optimal range (typically log P between 0 and 3 for oral drugs) is a primary objective in lead optimization [14] [57].
Gold Standard Experimental Techniques
Shake-Flask Method
The shake-flask method is widely regarded as the reference technique against which all other methods are validated due to its direct and intuitive approach to measuring partitioning behavior [59] [57].
Principle and Protocol
The method involves equilibrating the compound of interest between immiscible aqueous and organic phases, typically water-saturated n-octanol and n-octanol-saturated water [59] [14]. The flask is shaken vigorously to facilitate partitioning, allowed to stand for phase separation, and the concentration of the analyte is then quantified in one or both phases. The partition coefficient (P) or distribution coefficient (DpH) is calculated from the concentration ratio [59]. A key advantage of using Liquid Chromatography for quantification is its ability to separate impurities from the main component, ensuring analytical accuracy [59]. To manage a wide range of lipophilicities (-2 to 4.5 log D units) while using low drug amounts, optimized procedures employ different phase volume ratios [59].
Table 1: Key Steps in a Validated Shake-Flask Protocol for Low Drug Amounts [59]
| Step | Description | Key Considerations |
|---|---|---|
| 1. Phase Preparation | Prepare n-octanol saturated with aqueous buffer (e.g., pH 7.4 phosphate) and buffer saturated with n-octanol. | Ensures equilibrium and prevents phase volume changes during partitioning. |
| 2. Sample Preparation | Dissolve the drug in a small volume of DMSO or directly in one of the pre-saturated phases. | DMSO stocks are common in discovery; final DMSO concentration should be kept low (<1-2%). |
| 3. Equilibration | Combine the two phases in a vial and shake vigorously for a predetermined time. | Must be consistent to reach equilibrium without causing emulsion formation. |
| 4. Phase Separation | Allow the vial to stand or use centrifugation to achieve complete phase separation. | Critical for accurate sampling; micro-emulsions can complicate this step. |
| 5. Analysis | Quantify drug concentration in the aqueous phase (or both) using HPLC/UV. | Analyzing only the aqueous phase by difference is simpler and minimizes errors. |
| 6. Calculation | Calculate log D using the formula: log D = log [ (A_std / A_w) - 1 ] * (V_w / V_o ) where A=peak area, V=volume. |
Formula applied when analyzing only the aqueous phase and a standard [59]. |
The following workflow diagram illustrates the decision-making process for selecting the appropriate experimental technique based on the research objectives and compound characteristics.
Reversed-Phase Thin-Layer Chromatography (RP-TLC)
RP-TLC is a robust, simple, and cost-effective chromatographic technique endorsed by IUPAC for lipophilicity estimation [58] [60]. It offers advantages such as minimal solvent consumption, the ability to analyze several samples simultaneously, and no requirement for sophisticated instrumentation [60].
Principle and Protocol
In RP-TLC, the stationary phase is non-polar (e.g., silica gel impregnated with paraffin oil or chemically bonded C18 chains), and the mobile phase is a polar mixture, typically water with an organic modifier like methanol or acetone [58] [61]. The retention factor (Rf) is the primary data obtained, calculated as the distance migrated by the compound over the distance migrated by the solvent front [62]. To estimate lipophilicity, the Rf value is transformed into the RM value using the formula: RM = log (1/Rf - 1) [58]. Lipophilicity is then characterized by the RMW parameter, which is the theoretical RM value at 0% organic modifier, determined by extrapolating a linear plot of RM versus the concentration (φ) of the organic modifier [58] [60]. Studies have shown that the RMW parameter derived from Soczewiński–Wachtmeister’s equation can serve as an excellent alternative descriptor for the lipophilic nature of diverse drug classes [58].
Table 2: Common RP-TLC Systems for Lipophilicity Screening of Pharmaceuticals [58] [60]
| Drug Class (Example) | Stationary Phase | Mobile Phase (Organic Modifier : Water) | Lipophilicity Parameter | Key Finding |
|---|---|---|---|---|
| Antiparasitic & Antihypertensive | C18 bonded silica gel | Methanol-Water or Acetone-Water | RMW(S) (from Soczewiński–Wachtmeister’s equation) | Suitable for compounds with high and low lipophilicity [58]. |
| Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) | C18 bonded silica gel | Methanol-Water or Acetone-Water | RMW(O) (from Ościk’s equation) | More suitable for compounds with medium lipophilicity [58]. |
| β-Blockers | C18 bonded silica gel | Tetrahydrofuran (THF)-Water | C0 (equivalent to RMW) | C0 parameter from THF-water system was recommended for accurate lipophilicity estimation and protein binding prediction [60]. |
Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC)
RP-HPLC is a highly reproducible and efficient technique that is officially endorsed by the OECD for log P determination, particularly for compounds challenging to analyze via shake-flask [14].
Principle and Protocol
The principle is analogous to RP-TLC but with higher pressure and efficiency. The analyte is injected into a column packed with a non-polar stationary phase (e.g., C18) and eluted with a polar mobile phase. The retention time is used to calculate the capacity factor (k). The lipophilicity parameter is the log k value extrapolated to 0% organic modifier (log k0), or more commonly, the log kw obtained from the linear solvent-strength (LSS) model by plotting log k against the volume fraction of the organic modifier [14] [57]. This log kw value correlates strongly with log P and can be used to compare the relative lipophilicity of compounds within a series. The technique is highly automatable, making it ideal for medium- to high-throughput screening in drug discovery [14].
Comparative Analysis of Techniques
The choice between shake-flask, RP-TLC, and RP-HPLC depends on the research goals, available resources, and the nature of the compounds. The following table provides a structured comparison to guide this decision.
Table 3: Comparative Analysis of Gold Standard Lipophilicity Determination Methods
| Characteristic | Shake-Flask | RP-TLC | RP-HPLC |
|---|---|---|---|
| Principle | Direct partitioning between liquid phases [57]. | Indirect measurement via retention on a solid stationary phase [58] [60]. | Indirect measurement via retention on a solid stationary phase [14]. |
| Throughput | Low; labor-intensive and time-consuming [57]. | High; multiple samples run in parallel [60]. | Medium to High; automated and sequential analysis [14]. |
| Sample Purity | Requires pure compounds for accurate results. | Tolerates impurities; they are separated during development [60]. | Tolerates impurities; they are separated during elution [59]. |
| Sample Consumption | Relatively high [57]. | Low [60]. | Low [59] [57]. |
| Solvent Consumption | High (per sample). | Very Low (green chemistry) [14] [60]. | Medium to High. |
| Measured Parameter | log P or log D (direct) [14]. | RMW, C0 (chromatographic) [58] [60]. | log kw (chromatographic) [14]. |
| Typical Log P Range | -2 to 4 [59] [57]. | Wide range, depends on the system. | Wide range, depends on the system. |
| Key Advantage | Gold standard, direct measurement, physiologically intuitive [59] [57]. | Low cost, simplicity, high parallelism, green aspects [58] [60]. | High reproducibility, automation, OECD endorsed, high accuracy [14]. |
| Key Limitation | Low throughput, prone to emulsification, requires quantitative analysis [59] [57]. | Lower precision compared to HPLC, data processing can be less automated. | Requires more sophisticated instrumentation and method development. |
Correlation with ADMET Properties and Practical Applications
Chromatographically derived lipophilicity parameters are not merely theoretical substitutes; they have proven utility in predicting critical ADMET properties through Quantitative Structure-Retention Relationship (QSRR) and Quantitative Retention-Activity Relationship (QRAR) models.
For instance, in a study on β-blockers, a QRAR model was developed using the chromatographically determined lipophilicity parameter (C0) from a THF-water RP-TLC system and the maximal projection area of the molecule. This model allowed for the effective prediction of protein binding (PB), a key distribution parameter [60]. Similarly, RP-HPLC-derived log kw values are routinely used to model and predict passive membrane permeability, blood-brain barrier penetration, and other distribution-related processes [14] [57]. The strong correlation between chromatographic retention and biological partitioning arises from the analogous intermolecular forces at play (e.g., hydrophobic, van der Waals, hydrogen bonding) in both the chromatographic system and the lipid-aqueous interfaces in biological systems [14].
The following table lists essential reagents and materials commonly employed in these experimental techniques.
Table 4: Essential Research Reagent Solutions for Lipophilicity Assays
| Reagent/Material | Typical Application | Function in the Experiment |
|---|---|---|
| n-Octanol (water-saturated) | Shake-Flask Method | Organic phase in the partitioning system, mimicking lipid environments [59] [14]. |
| Aqueous Buffer (e.g., Phosphate, pH 7.4) | Shake-Flask Method; RP-HPLC | Aqueous phase for partitioning; simulates physiological pH for log D7.4 determination [59] [14]. |
| C18-Bonded Silica Plates | RP-TLC | Serves as the non-polar (reversed-phase) stationary phase [58] [60]. |
| C18-Bonded Silica Columns | RP-HPLC | Serves as the non-polar (reversed-phase) stationary phase for high-efficiency separation [14]. |
| Organic Modifiers (Methanol, Acetonitrile, Tetrahydrofuran) | RP-TLC & RP-HPLC | Component of the mobile phase to modulate retention and elution strength [58] [60]. |
| Polyoxyethylene (23) lauryl ether (Brij 35) | Biopartitioning Micellar Chromatography (BMC) | Forms micelles in the mobile phase to better mimic biological membrane barriers [60]. |
The shake-flask, RP-TLC, and RP-HPLC techniques represent the experimental backbone for reliable lipophilicity assessment in drug discovery. While the shake-flask method remains the definitive gold standard for direct measurement, chromatographic techniques offer superior throughput, reproducibility, and practical utility for screening compound libraries. The choice of technique is strategic: shake-flask for validation, RP-HPLC for efficient and robust screening, and RP-TLC for a cost-effective and green alternative, especially in early-stage discovery or resource-limited settings. Critically, the lipophilicity parameters obtained from these methods are not mere numbers but are indispensable for constructing predictive models of ADMET behavior. By integrating these experimental tools with modern in silico approaches, researchers can more effectively navigate the complex landscape of drug design, de-risking the development process and enhancing the likelihood of creating successful therapeutic agents with optimal pharmacokinetic profiles.
In modern drug discovery, the high attrition rates of candidate compounds due to unfavorable pharmacokinetics and toxicity profiles have underscored the critical importance of early-stage absorption, distribution, metabolism, excretion, and toxicity (ADMET) evaluation. Among various physicochemical parameters, lipophilicity serves as a principal determinant of ADMET behavior, influencing membrane permeability, protein binding, metabolic stability, and volume of distribution [63] [64]. Computational (in silico) prediction platforms have emerged as indispensable tools for profiling compound properties prior to costly synthesis and experimental testing [65]. This technical guide provides a comprehensive comparative analysis of three prominent in silico platforms—SwissADME, pkCSM, and StarDrop—with particular emphasis on their application in lipophilicity-informed ADMET optimization. Each platform offers distinct methodologies, capabilities, and implementation frameworks, enabling researchers to select appropriate tools based on specific project requirements within the context of rational drug design.
The Critical Role of Lipophilicity in ADMET Properties
Lipophilicity, commonly quantified as the partition coefficient between n-octanol and water (Log P), exerts multifaceted influences on drug disposition and kinetics. A thorough understanding of these relationships is fundamental to interpreting in silico predictions and designing compounds with optimal pharmacokinetic profiles.
Absorption and Permeation: Sufficient lipophilicity enables compounds to traverse biological membranes such as the gastrointestinal mucosa, but excessive lipophilicity can impair aqueous solubility and dissolution, thereby limiting absorption [63]. This balance is crucial for achieving adequate oral bioavailability.
Tissue Distribution and Protein Binding: Lipophilicity strongly correlates with the volume of distribution (Vd), as demonstrated by studies showing Vd increases of 0.25-0.66 L/kg in obese patients compared to normal-weight controls for highly lipophilic drugs like imipramine and verapamil [64]. This relationship arises from enhanced partitioning into adipose tissue and cellular membranes, which can both improve target site delivery and promote off-target accumulation.
Metabolic Clearance and Half-Life: Highly lipophilic compounds typically exhibit greater access to hepatic metabolic enzymes, potentially accelerating clearance. However, the dominant effect of lipophilicity often manifests through distribution rather than clearance mechanisms. Increased Vd directly prolongs elimination half-life, creating potential challenges for drug accumulation and washout, particularly in special populations such as obese patients [64].
Molecular Design Considerations: While increasing lipophilicity often enhances target affinity due to the hydrophobic nature of many binding pockets, this strategy carries diminishing returns. Excessive lipophilicity (Log P >5) frequently correlates with poor solubility, promiscuous binding, and heightened toxicity risk [63]. Modern drug discovery has witnessed a gradual increase in average Log P values of approved drugs, primarily driven by declining proportions of highly polar molecules rather than increases in extremely lipophilic compounds [63].
Table 1: Key Lipophilicity Parameters and Their ADMET Influences
| Parameter | Definition | Primary ADMET Influences |
|---|---|---|
| Log P | Partition coefficient of neutral form between n-octanol and water | Membrane permeability, metabolic stability, volume of distribution |
| Log D | Distribution coefficient at specific pH (typically 7.4) | Ionization-dependent partitioning, absorption pH-dependence |
| Chromatographic Hydrophobicity | HPLC retention time as lipophilicity indicator | Correlates with in vivo volume of distribution [64] |
SwissADME
SwissADME represents a freely accessible web tool that integrates robust predictive models for physicochemical properties, pharmacokinetics, drug-likeness, and medicinal chemistry friendliness [66]. Its development was motivated by the need to consolidate diverse ADMET prediction capabilities within a single, academically available platform. The tool employs multiple calculation approaches for key parameters, particularly for lipophilicity estimation where it provides five distinct prediction methods (iLOGP, XLOGP3, WLOGP, MLOGP, SILICOS-IT) with a consensus value to balance methodological strengths and limitations [66] [67]. This multi-algorithm approach addresses the well-documented challenge of prediction variance across different chemical series.
The platform features several proprietary models including the BOILED-Egg (Brain Or IntestinaL EstimateD permeation) graphical model for predicting gastrointestinal absorption and blood-brain barrier penetration, and the Bioavailability Radar for rapid assessment of drug-likeness across six key physicochemical parameters [66]. For experimental protocols, SwissADME requires researchers to input molecular structures as SMILES notations, preferably in their neutral form to ensure prediction accuracy, as models are trained primarily on neutral compounds [67]. The platform efficiently processes batch submissions of up to 200 molecules simultaneously, with computation times of approximately 1-5 seconds per drug-like molecule [67].
pkCSM
The pkCSM platform employs a distinctive graph-based signature approach that encodes distance patterns between atoms to represent molecular structure and train predictive models for pharmacokinetic properties [68]. This methodology captures complex structural relationships that may not be evident from conventional descriptors, potentially offering enhanced predictive accuracy for challenging ADMET endpoints. The platform encompasses five primary pharmacokinetic property classes, utilizing both regression and classification approaches tailored to specific prediction tasks.
As a web-based tool, pkCSM offers accessibility advantages for academic researchers, with no license requirements for non-commercial applications [68]. While detailed methodological specifications are available in the platform's primary literature, its prediction of lipophilicity-dependent parameters aligns with the fundamental relationships between molecular structure, hydrophobic interactions, and biological disposition. The platform's graph-based framework may offer particular value for predicting complex distribution and metabolism phenomena where traditional descriptors prove insufficient.
StarDrop
StarDrop represents a comprehensive commercial drug discovery suite with specialized modules for ADMET prediction, including the optional ADME QSAR module which provides high-quality quantitative structure-activity relationship models for a broad spectrum of properties [69]. Unlike the freely accessible platforms, StarDrop employs a commercial licensing model with annual subscriptions typically starting at approximately $10,000, reflecting its extensive capabilities and integration features [70].
The platform's distinctive strength lies in its implementation of confidence estimates for every prediction, clearly identifying when molecules fall outside the applicable chemical space where model reliability may be compromised [69]. This transparency in uncertainty assessment represents a critical feature for informed decision-making in lead optimization. StarDrop's ADME QSAR module predicts an extensive range of lipophilicity-influenced parameters including log P, log D at pH 7.4, cytochrome P450 affinities, plasma protein binding, P-gp transport, and human intestinal absorption [69]. The platform further supports custom QSAR model development through its Auto-Modeller component, enabling organizations to incorporate proprietary data and refine predictions for specific chemical series [70].
Comparative Analysis of Platform Capabilities
Table 2: Platform Comparison: Access, Lipophilicity Prediction, and Key Features
| Feature | SwissADME | pkCSM | StarDrop |
|---|---|---|---|
| Access Model | Free web tool [66] | Free for academia [68] | Commercial ($10,000+ annually) [70] |
| Lipophilicity Prediction | Five methods + consensus [66] | Graph-based signatures [68] | QSAR models with confidence estimation [69] |
| Key Lipophilicity-Dependent Predictions | BOILED-Egg, bioavailability radar, drug-likeness filters [66] | Broad pharmacokinetic properties [68] | logP, logD, CYP affinities, PPB, P-gp, HIA [69] |
| Throughput | Batch processing (≤200 molecules) [67] | Not specified | High-throughput compatible [70] |
| Special Features | Integrated with SwissDrugDesign tools [66] | Novel graph-based structural signatures [68] | Confidence estimation, custom model development [69] |
Lipophilicity Prediction Methodologies
SwissADME: Employs a diverse ensemble of lipophilicity estimation methods including iLOGP (a physics-based method using free energies of solvation), XLOGP3 (an atomistic method with corrective factors), WLOGP (an atomistic fragmental system), MLOGP (a topological method), and SILICOS-IT (a hybrid approach) [66]. This methodological diversity acknowledges the well-established limitation that no single approach consistently outperforms others across diverse chemical spaces [67]. The platform's consensus log P value represents an arithmetic mean of these predictions, providing a balanced estimate that mitigates individual methodological biases.
pkCSM: Utilizes graph-based signatures that encode distance patterns between atoms to represent molecular structure [68]. This approach captures complex topological relationships that may correlate with lipophilicity and related ADMET behaviors, potentially identifying structure-property relationships overlooked by conventional descriptors.
StarDrop: Implements QSAR models trained on extensive experimental datasets, with applicability domain assessment that identifies when query compounds diverge from the training chemical space [69] [71]. This confidence estimation critically informs interpretation of lipophilicity-dependent predictions, particularly for novel chemotypes where model extrapolation may be unreliable.
ADMET Endpoints with Lipophilicity Dependence
Each platform predicts numerous ADMET endpoints where lipophilicity serves as a key determinant:
SwissADME: Provides predictions for P-glycoprotein substrate status, CYP450 inhibition, and skin permeation (Log Kp), all exhibiting strong lipophilicity dependence [66] [67]. The platform's BOILED-Egg model directly visualizes the interplay between lipophilicity and polarity in determining brain access and intestinal absorption [66].
pkCSM: Predicts a comprehensive range of pharmacokinetic properties across five main classes using its graph-based signature approach [68]. While specific endpoints aren't detailed in the available sources, the methodology is reportedly applicable to fundamental ADMET behaviors including distribution, metabolism, and excretion processes with documented lipophilicity relationships.
StarDrop: Offers an extensive suite of QSAR models for lipophilicity-dependent parameters including log P, log D, plasma protein binding, cytochrome P450 affinities (CYP2C9, CYP2D6), P-gp transport, human intestinal absorption, and blood-brain barrier penetration [69]. The platform's emphasis on prediction confidence is particularly valuable for these endpoints, where lipophilicity relationships may display significant nonlinearity and context dependence.
Practical Implementation and Workflow Integration
Experimental Protocols for In Silico Screening
Researchers can implement the following standardized protocol for comprehensive lipophilicity and ADMET assessment:
Compound Preparation and Input
- Generate accurate SMILES notations for all query compounds, ensuring representation in their neutral states (except permanent ions or zwitterions) [67].
- For batch processing, prepare a text file with one SMILES entry per line, optionally including compound identifiers separated by spaces [67].
- Verify structural integrity through visual inspection of rendered structures in platform interfaces.
Platform-Specific Execution
- SwissADME: Input structures via the Marvin JS sketcher or direct SMILES entry. Initiate batch processing for collections of up to 200 compounds. Download results in CSV format for further analysis [66] [67].
- pkCSM: Submit structures through the web interface using the graph-based signature methodology. Retrieve predictions for the comprehensive set of pharmacokinetic endpoints [68].
- StarDrop: Import compound structures through the interactive interface. Execute ADME QSAR models with particular attention to confidence indicators for each prediction [69] [70].
Lipophilicity Data Interpretation
- Compare multiple prediction values for log P where available (especially in SwissADME) to assess consensus and identify potential outliers [67].
- Evaluate lipophilicity-dependent parameters (e.g., permeability, distribution, metabolism) in the context of established optimal ranges for the intended route of administration and therapeutic target [63] [64].
- Utilize visualization tools such as the Bioavailability Radar in SwissADME to rapidly identify deviations from optimal physicochemical space [66].
Decision-Making and Optimization
- Prioritize compounds exhibiting balanced lipophilicity profiles (typically Log P 1-3) with favorable predicted ADMET characteristics [63].
- Identify structural features associated with undesirable lipophilicity-driven properties (e.g., excessive metabolic lability, P-gp substrate behavior) for subsequent optimization.
- Iterate the design-predict-analyze cycle to refine lead compounds while maintaining lipophilicity within the optimal range for the specific therapeutic application.
Essential Research Reagent Solutions
Table 3: Key In Silico Research Tools for Lipophilicity-ADMET Studies
| Tool Category | Representative Examples | Research Application |
|---|---|---|
| Molecular Descriptors | Topological Polar Surface Area (TPSA), Molecular Refractivity, H-bond Donor/Acceptor Counts [66] | Quantification of physicochemical properties influencing lipophilicity and ADMET |
| Drug-likeness Filters | Lipinski's Rule of Five, Ghose Filter, Veber Rule [66] | Rapid assessment of developability based on lipophilicity and related parameters |
| QSAR Model Builders | StarDrop Auto-Modeller [70] | Development of customized prediction models for proprietary chemical series |
| Visualization Tools | BOILED-Egg Model [66], Bioavailability Radar [66] | Intuitive interpretation of absorption and distribution potential |
SwissADME, pkCSM, and StarDrop offer complementary approaches to in silico ADMET prediction with particular utility for profiling lipophilicity-dependent properties. SwissADME provides an exceptional accessibility-feature balance for academic researchers and preliminary screening. pkCSM introduces innovative graph-based methodologies that may capture complex structure-property relationships. StarDrop delivers enterprise-grade solutions with confidence estimation and customization capabilities suitable for advanced drug discovery programs. Informed selection and application of these platforms, grounded in a thorough understanding of lipophilicity-ADMET relationships, can significantly enhance efficiency in early drug discovery by prioritizing compounds with optimal physicochemical and pharmacokinetic profiles.
Lipophilicity is a fundamental physicochemical parameter defined as the affinity of a molecule for a lipophilic environment versus an aqueous one [37]. It is most commonly quantified as the decimal logarithm of the partition coefficient (Log P) in an n-octanol/water system, which serves as a model for the partitioning of drugs between aqueous biological fluids and lipid membranes [37]. For ionizable compounds, the distribution coefficient (Log D) at a physiologically relevant pH, such as 7.4, provides a more meaningful description of lipophilicity [37]. This property is a critical determinant in drug discovery and development because it profoundly influences a compound's absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile [72] [73] [11]. Moderately lipophilic compounds generally demonstrate better absorption and membrane permeability, while excessive lipophilicity can lead to poor aqueous solubility, increased metabolic clearance, and higher risk of toxicity [74].
1,9-Diazaphenothiazines are a group of modified phenothiazines in which two benzene rings of the classic phenothiazine structure have been replaced with pyridine rings [75]. These compounds have recently attracted significant research interest due to their promising and impressive in vitro anticancer potential, which is associated with the activation of the mitochondrial apoptosis pathway [72] [75]. This activity is connected with the induction of BAX, forming a channel in the mitochondrial outer membrane, and the subsequent release of cytochrome c for the activation of caspases 9 and 3 [72]. The study of their lipophilicity is therefore essential for understanding their bioavailability and optimizing their pharmacokinetic properties. This case study details the experimental and in silico strategies employed to determine the lipophilicity of 1,9-diazaphenothiazines and explores its impact on their ADMET properties.
Experimental Determination of Lipophilicity
Reversed-Phase Thin-Layer Chromatography (RP-TLC)
The lipophilicity of 1,9-diazaphenothiazines was determined experimentally using the Reversed-Phase Thin-Layer Chromatography (RP-TLC) method [72] [73]. This technique is an indirect method that has largely replaced the classical shake-flask technique for lipophilicity screening due to its higher throughput, lower consumption of samples and solvents, and good reproducibility [11].
Detailed Methodology:
- Stationary Phase: RP-TLC plates pre-coated with modified silica gel RP-18 F254S were used as the non-polar phase [73].
- Mobile Phase: A mixture of acetone and TRIS buffer (0.2 M, pH = 7.4) was employed as the polar mobile phase to mimic physiological conditions [73]. The mobile phase was prepared in different volume ratios, typically ranging from 50:50 to 70:30 (acetone:TRIS buffer) [73].
- Application and Development: Solutions of the tested 1,9-diazaphenothiazine compounds and a set of standard compounds with known literature Log P (log Plit.) values were applied to the plate. The plate was then developed in a chromatographic chamber pre-saturated with the vapor of the mobile phase [73].
- Detection and Calculation: After development, the spots were visualized under UV light at a wavelength of 254 nm. The retention factor (RF) was calculated for each compound. The RM value was then determined using the formula RM = log(1/RF - 1) [73]. A graph of RM versus the concentration (C) of acetone in the mobile phase was plotted for each compound. The relative lipophilicity parameter (RM0), which is an indicator of the partition between the non-polar stationary phase and the polar mobile phase, was obtained by extrapolating the line to zero acetone concentration, according to the equation RM = RM0 + bC, where b represents the specific hydrophobic surface area [73].
- Conversion to log PTLC: The RM0 values were subsequently transformed into the partition coefficient (log PTLC) using a calibration curve constructed from the standard compounds [73].
The workflow for this experimental determination is outlined in the diagram below.
The Shake-Flask Method and Alternatives
The shake-flask method is considered the gold standard for the direct experimental determination of the partition coefficient and is recommended by the Organization for Economic Co-operation and Development (OECD) [76] [11]. It involves dissolving the compound in a biphasic system of n-octanol and water, shaking the mixture vigorously until equilibrium is reached, and then measuring the concentration of the compound in each phase [76]. While highly accurate for Log P values between -2 and 4, the method is labor-intensive, time-consuming (requiring 1 to 24 hours to reach equilibrium), and requires relatively large amounts of pure compound [76] [11].
Several modern variations have been developed to address these limitations:
- Slow-Stirring Method: This method uses slow stirring instead of vigorous shaking to prevent the formation of emulsions, which is particularly useful for compounds with very high lipophilicity (Log P > 4.5) [76].
- Vortex-Assisted Liquid-Liquid Microextraction (VALLME): This method uses vortex agitation to disperse microvolumes of n-octanol in the aqueous phase, creating a large surface area for partitioning and dramatically reducing the equilibrium time to approximately 2 minutes [76].
- Water-Plug Aspiration/Injection Method: This technique is designed to minimize phase contamination during sampling, which is a common issue when dealing with the high viscosity of n-octanol, making it suitable for highly lipophilic compounds [76].
Research Reagent Solutions
The following table details key reagents and materials used in the experimental determination of lipophilicity for 1,9-diazaphenothiazines.
Table 1: Essential Research Reagents for Lipophilicity Determination
| Reagent/Material | Function in the Experiment |
|---|---|
| RP-18 F254S TLC Plates | The stationary phase; provides a non-polar surface for compound partitioning [73]. |
| n-Octanol and Water | The biphasic solvent system for the shake-flask method, representing the lipid and aqueous phases, respectively [76] [37]. |
| TRIS Buffer (pH 7.4) | Provides a stable, physiologically relevant pH for the mobile phase in RP-TLC or for the aqueous phase in shake-flask, determining the ionization state of the compound [73]. |
| Acetone | Organic modifier in the RP-TLC mobile phase; its concentration is varied to create the correlation for RM0 determination [73]. |
| Standard Compounds (e.g., benzamide, benzophenone) | Compounds with known literature Log P values used to construct the calibration curve for converting RM0 to log PTLC [73]. |
In Silico Prediction and Data Integration
Computational Methods
In silico methods are indispensable in modern drug discovery for the rapid prediction of lipophilicity, especially during the early stages of development [76] [73] [11]. These methods estimate the partition coefficient (log Pcalcd.) from the molecular structure using various algorithms. For 1,9-diazaphenothiazines, calculations were performed using multiple programs and mathematical models available on web servers such as VCCLAB, SwissADME, and ChemDraw [73]. The models employed include iLOGP, XLOGP3, WLOGP, MLOGP, SILICOS-IT, and others [72] [73]. However, calculated values should be considered estimates, as they can vary by more than two log P units depending on the algorithm used [76] [73]. They are highly useful for filtering drug-like compounds and selecting experimental conditions, but they should be verified with experimental data as soon as possible [76].
Lipophilicity Data for 1,9-Diazaphenothiazines
The experimental and computational studies confirmed that 1,9-diazaphenothiazines possess moderate lipophilicity. The experimental log PTLC values for this class of compounds were found to be in a range that supports good bioavailability [72]. The following table summarizes the types of lipophilicity data obtained for these compounds.
Table 2: Experimentally Determined and Theoretically Calculated Lipophilicity Parameters of 1,9-Diazaphenothiazines
| Parameter | Description | Typical Finding for 1,9-Diazaphenothiazines |
|---|---|---|
| RM0 | Chromatographic parameter representing relative lipophilicity from RP-TLC. | Determined for each derivative; used for comparison and conversion [72] [73]. |
| log PTLC | Partition coefficient derived from RM0 via a calibration curve. | Reported values indicate moderate lipophilicity, favorable for drug absorption [72]. |
| log Pcalcd. | Computed partition coefficient from various software. | Values varied between algorithms but generally aligned with the moderate lipophilicity trend [72] [73]. |
| b (slope) | Specific hydrophobic surface area from the RM vs. C plot. | Provided information on the congeneric nature of the tested compounds [73]. |
Correlation of Lipophilicity with ADMET Properties
The lipophilicity of 1,9-diazaphenothiazines has a direct impact on their ADMET properties, which were investigated in silico using servers like SwissADME and SwissTargetPrediction [72].
- Absorption and Distribution: Moderate lipophilicity enables a compound to dissolve in gastrointestinal fluids and passively diffuse across lipid membranes, facilitating absorption [74]. Furthermore, lipophilicity is a principal correlate of a drug's volume of distribution (Vd). Higher lipophilicity generally leads to a larger Vd, as drugs are more likely to distribute from the plasma into tissues and lipid depots [64]. This effect can be disproportionately greater in obese patients for highly lipophilic drugs, significantly prolonging their elimination half-life [64].
- Metabolism and Excretion: Highly lipophilic compounds are more susceptible to metabolism by hepatic enzymes (e.g., via oxidation), which can shorten their half-life [64] [74]. They may also be reabsorbed in the kidneys, leading to slower excretion [37].
- Toxicity: Optimizing lipophilicity helps avoid off-target effects and reduces the risk of accumulation in lipid tissues, which could lead to chronic toxicity [74].
- Drug-likeness: The tested 1,9-diazaphenothiazines were found to comply with Lipinski's Rule of Five, as well as Ghose's and Veber's rules, confirming their potential for good oral bioavailability [72]. This is a direct consequence of their well-balanced lipophilicity and other physicochemical properties.
The central role of lipophilicity in the activity of 1,9-diazaphenothiazines is summarized in the pathway below.
This case study demonstrates a comprehensive workflow for determining the lipophilicity of anticancer 1,9-diazaphenothiazines. The integrated approach, combining experimental RP-TLC methodology with in silico predictions, provides a robust strategy for profiling this critical physicochemical parameter. The findings confirm that 1,9-diazaphenothiazines possess moderate lipophilicity, which is a key factor contributing to their favorable ADMET profile and strong in vitro anticancer activity, linked to the induction of the mitochondrial apoptosis pathway. The successful application of these techniques underscores their vital role in modern anticancer drug development, enabling researchers to optimize lead compounds for desired pharmacokinetic and pharmacodynamic outcomes.
Lipophilicity is a fundamental physicochemical property that significantly influences the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of potential drug candidates [14]. Defined as the partition coefficient (logP) of a compound between n-octanol and water, lipophilicity serves as a critical descriptor for predicting a molecule's behavior in biological systems [77]. In modern drug discovery, poor ADMET characteristics account for approximately 60% of drug failures in clinical phases, highlighting the importance of early evaluation of these properties [78]. This case study examines the lipophilicity and ADMET parameters of novel anticancer diquinothiazine hybrids within the broader context of optimizing pharmacokinetic profiles during early drug development stages.
Diquinothiazines represent a promising class of pentacyclic phenothiazine derivatives with demonstrated anticancer activity [15]. These nitrogen- and sulfur-containing heterocyclic compounds have shown significant antiproliferative effects against various cancer cell lines, including glioblastoma SNB-19, colorectal carcinoma Caco-2, breast cancer MDA-MB-231, and lung cancer A549 cells [15]. The most active compounds in this series, particularly dimethylaminopropyldiquinothiazine and pyrrolidinylethyldiquinothiazine, have exhibited IC50 values as low as 0.3 μM, surpassing the activity of cisplatin in certain cell lines [15]. Understanding the lipophilicity and ADMET profiles of these promising compounds provides crucial insights for their further development as potential therapeutic agents.
Experimental and Computational Methodologies
Chromatographic Determination of Lipophilicity
The experimental lipophilicity of diquinothiazine hybrids was determined using reversed-phase thin-layer chromatography (RP-TLC) with RP-18 silica gel plates as the stationary phase [15]. The mobile phase consisted of acetone-TRIS buffer (0.2 M, pH 7.4) mixtures with acetone concentrations varying from 50% to 80% [78]. This method offers several advantages over traditional shake-flask techniques, including reduced sample requirements, faster analysis times, and improved suitability for compounds with limited solubility [77].
The chromatographic procedure involved applying 5 μL of ethanolic solutions of each diquinothiazine hybrid to RP-TLC plates, with development in equilibrated chambers at room temperature [78]. After development, compound spots were visualized using iodine vapor. The retardation factor (Rf) values were converted to RM values using the equation RM = log(1/Rf - 1) [78]. The lipophilicity parameter (RM0) was determined by extrapolating the RM values to zero organic modifier concentration using the equation RM = RM0 + bC, where C represents the concentration of acetone in the mobile phase [15].
For absolute lipophilicity values, the relative RM0 parameters were converted to logPTLC values using a calibration curve constructed with reference compounds of known lipophilicity: benzamide (A), acetanilide (B), 4-bromoacetophenone (C), benzophenone (D), anthracene (E), and DDT (F), covering a logP range of 0.64-6.38 [78]. The resulting calibration equation was logPTLC = 1.1405 RM0 - 0.0787 (r = 0.999; SD = 0.102) [78].
Computational Prediction Approaches
Computational methods for lipophilicity prediction employed multiple algorithms to estimate partition coefficients, providing complementary data to experimental measurements [15]. The study utilized various software tools including iLOGP, XLOGP3, WLOGP, MLOGP, SILCOS-IT, and milogP, which employ different theoretical methodologies such as atomic approaches, piecewise contribution techniques, and property-dependent methods [15] [77].
ADMET parameters were predicted in silico using two primary platforms: SwissADME and pkCSM [15]. These tools evaluate drug-likeness based on Lipinski's "Rule of Five," which states that compounds are more likely to have good oral bioavailability when they have: MlogP ≤ 4.15, molecular weight ≤ 500 g/mol, hydrogen bond donors ≤ 5, and hydrogen bond acceptors ≤ 10 [79]. Additional ADMET risk assessment was performed using specialized software that calculates overall risk scores based on absorption, CYP metabolism, and toxicity parameters [79].
Table 1: Key Experimental and Computational Methods for Lipophilicity and ADMET Assessment
| Method Category | Specific Technique/Software | Primary Application | Key Advantages |
|---|---|---|---|
| Experimental Lipophilicity | RP-TLC (RP-18 plates) | Determination of RM0 and logPTLC values | Minimal sample requirement, rapid analysis, suitable for insoluble compounds |
| Computational Lipophilicity | iLOGP, XLOGP3, WLOGP, MLOGP | Prediction of partition coefficients | Rapid screening, applicable prior to synthesis |
| ADMET Prediction | SwissADME, pkCSM | Comprehensive ADMET profiling | Early risk assessment, cost-effective screening |
| ADMET Risk Assessment | ADMET Predictor modules | Integrated risk scoring for absorption, CYP metabolism, and toxicity | Mechanistic insights, quantitative risk evaluation |
Figure 1: Workflow for Lipophilicity and ADMET Profiling of Diquinothiazine Hybrids. The integrated approach combines experimental and computational methods for comprehensive pharmacokinetic assessment.
Results and Discussion
Lipophilicity Parameters of Diquinothiazine Hybrids
The experimental lipophilicity study revealed that diquinothiazine hybrids generally exhibited moderate lipophilicity, with RM0 values varying based on their structural features, particularly the nature and position of substituents [15]. The chromatographic parameter RM0 showed good correlation with computationally derived logP values, though the degree of correlation varied depending on the specific algorithm used [15]. Among all computational methods evaluated, iLOGP demonstrated the closest similarity to chromatographically determined lipophilicity (RM0) for certain diquinothiazine compounds [15].
Comparative analysis of different diquinothiazine isomers indicated that most computational programs did not distinguish between structural isomers, with AClogP, ALOGP, XLOGP2, and XLOGP3 calculating identical logPcalcd values for all isomeric diquinothiazines [77]. However, the ALOGPS program successfully differentiated between isomers, calculating logPcalcd values of 4.90 and 4.92 for diquinothiazines 1 and 9, respectively, while assigning values of 4.93 for diquinothiazines 5 and 11 [77]. The MLOGP program also distinguished derivative 1 (logPcalcd = 2.78) from the other three structural isomers (logPcalcd = 3.17) [77].
The lipophilicity values observed for diquinothiazine hybrids generally fell within the optimal range for drug-like compounds according to Lipinski's rule, which recommends logP values below 5 for good oral bioavailability [14]. Compounds with moderate lipophilicity (logP 0-3) typically demonstrate balanced solubility and permeability characteristics, making them suitable candidates for oral administration [14].
Table 2: Lipophilicity Determination Methods and Their Applications in Drug Discovery
| Method Type | Specific Technique | Key Parameters | Optimal Range for Drug Candidates | Limitations |
|---|---|---|---|---|
| Chromatographic | RP-TLC | RM0, logPTLC | logP 0-3 (oral drugs) | Requires reference compounds for calibration |
| Chromatographic | RP-HPLC | logk0 | logP ~2 (BBB penetration) | Method development can be time-consuming |
| Computational | iLOGP, XLOGP3 | logPcalcd | logP < 5 (Lipinski's Rule) | Algorithm-dependent variability |
| Classical | Shake-flask | logP | -2 to 4 (reliable range) | Time-consuming, requires pure compounds |
ADMET Profiling and Drug-Likeness Evaluation
In silico ADMET prediction using SwissADME and pkCSM platforms provided comprehensive pharmacokinetic profiles of the diquinothiazine hybrids [15]. The results indicated that most tested compounds complied with the rules of Lipinski, Veber, and Egan, suggesting strong potential for development as orally active therapeutic agents, particularly for non-CNS targets [77]. Specific ADMET parameters analyzed included Caco-2 permeability, P-glycoprotein inhibition potential, and interactions with cytochrome P450 enzymes, particularly CYP2C19 and CYP3A4 isoforms [77].
The correlation analysis between experimental lipophilicity (logPTLC) and various ADMET parameters revealed generally poor predictive power for specific endpoints such as Caco-2 permeability, P-gp inhibition, and CYP enzyme interactions [77]. This finding underscores the complexity of ADMET properties and suggests that lipophilicity alone is insufficient for accurate prediction of these parameters, necessitating comprehensive in silico profiling using specialized platforms.
Toxicity risk assessment indicated that the diquinothiazine hybrids showed promising safety profiles, with no significant neurotoxicity concerns identified through in silico evaluation [15]. This is particularly important for anticancer agents intended for chronic administration, where toxicity considerations often limit clinical utility. The integration of ADMET risk assessment, which evaluates absorption, CYP metabolism, and toxicity parameters using soft threshold ranges, provided quantitative risk scores for prioritizing lead compounds [79].
Figure 2: Impact of Lipophilicity on Key ADMET Properties. Lipophilicity influences all aspects of a drug's pharmacokinetic profile, from absorption to toxicity.
Structural Considerations and Property Optimization
The structural features of diquinothiazine hybrids significantly influenced both their lipophilicity and ADMET parameters. Introduction of dialkylaminoalkyl substituents at specific positions of the diquinothiazine core enabled fine-tuning of lipophilicity to achieve optimal values for drug-like properties [15]. The nature of the substituents played a critical role in determining the overall pharmacokinetic profile, with smaller alkyl groups generally reducing lipophilicity compared to bulkier aromatic substituents.
The method of ring condensation in the pentacyclic system also impacted physicochemical properties. Angularly condensed diquinothiazines and quinonaphthothiazines demonstrated distinct lipophilicity profiles compared to their linearly fused counterparts, highlighting the importance of molecular topology in property-based design [77]. These structural modifications affected not only lipophilicity but also molecular geometry, hydrogen bonding capacity, and electronic distribution, all of which contribute to overall drug-like characteristics.
Analysis of global reactivity descriptors based on density functional theory (DFT) calculations provided additional insights into the electronic properties of diquinothiazine hybrids and their potential for interactions with biological targets [78]. Frontier molecular orbital (HOMO-LUMO) energies and molecular electrostatic potentials correlated with the observed biological activities and binding interactions identified through molecular docking studies [80].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Materials for Lipophilicity and ADMET Studies
| Reagent/Material | Specification | Primary Function | Application Notes |
|---|---|---|---|
| RP-TLC Plates | Silica gel RP-18 F254 | Stationary phase for chromatographic lipophilicity determination | Enable high-resolution separation of compounds based on hydrophobicity |
| Mobile Phase | Acetone-TRIS buffer (pH 7.4) | Liquid phase for RP-TLC analysis | TRIS buffer mimics physiological pH; acetone as organic modifier |
| Reference Compounds | Benzamide, acetanilide, 4-bromoacetophenone, benzophenone, anthracene, DDT | Calibration standards for logPTLC determination | Cover broad logP range (0.64-6.38) for accurate calibration |
| Visualization Reagent | Iodine vapor | Detection of compound spots on TLC plates | Non-destructive method allowing further analysis |
| Software - SwissADME | Web tool | Comprehensive ADMET prediction | Evaluates drug-likeness, pharmacokinetics, and medicinal chemistry friendliness |
| Software - pkCSM | Web platform | ADMET property prediction | Provides parameters for permeability, metabolism, toxicity, and distribution |
| Software - ADMET Predictor | Commercial platform with >175 models | Advanced ADMET risk assessment | Machine learning-based predictions with applicability domain assessment |
This case study demonstrates the critical importance of integrated lipophilicity assessment and ADMET profiling in the development of novel diquinothiazine hybrids as anticancer agents. The combination of chromatographic techniques and computational methods provides a robust framework for evaluating these essential physicochemical and pharmacokinetic parameters during early drug discovery stages.
The findings confirm that diquinothiazine hybrids generally exhibit favorable drug-like properties, with moderate lipophilicity values that support potential oral bioavailability. The successful application of RP-TLC for experimental lipophilicity determination, coupled with in silico predictions using platforms such as SwissADME and pkCSM, offers a cost-effective strategy for prioritizing lead compounds with optimal ADMET characteristics.
Future directions in this field include the expanded implementation of machine learning models for ADMET prediction, which have demonstrated significant promise in predicting key endpoints such as solubility, permeability, metabolism, and toxicity [81]. These approaches, combined with experimental validation, hold potential for further accelerating the development of diquinothiazine-based therapeutics with optimized pharmacokinetic profiles and enhanced clinical translation potential.
The journey of a drug candidate from administration to its site of action is governed by its fundamental physicochemical properties. Among these, lipophilicity stands as a primary underlying structural property that affects higher-level biochemical and pharmacokinetic behaviors [25]. However, its influence cannot be viewed in isolation; it is part of an interdependent system that includes topological polar surface area (TPSA), hydrogen bond donors (HBD), and molecular weight (MW). These properties collectively form the foundation of modern drug design principles, such as the renowned Rule of Five (Ro5), which serves as an initial indicator of a compound's likelihood of achieving oral bioavailability [82]. This guide examines the integrated roles of these critical descriptors within the context of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) research, providing a technical framework for their application in rational drug design.
Defining the Core Descriptors
Lipophilicity (LogP/LogD)
Lipophilicity, quantified as the partition coefficient (LogP) or its pH-dependent distribution coefficient (LogD), measures a molecule's affinity for a lipophilic environment relative to an aqueous one [83]. It is arguably the single most important physical property affecting a drug's potency, distribution, and elimination [25].
- Measurement and Calculation: The classic shake-flask method, recommended by the OECD, directly measures the partition coefficient but is time-consuming and requires pure compounds in relatively large amounts [15]. Chromatographic techniques like RP-TLC and RP-HPLC offer faster, indirect determinations of lipophilicity parameters (e.g., RM0) with high repeatability [15]. Numerous in silico tools are available for rapid prediction, including atomistic methods (XLOGP3, WLOGP), topological methods (MLOGP), and physics-based methods (iLOGP). A consensus log Po/w from multiple predictors often increases accuracy [66].
Topological Polar Surface Area (TPSA)
TPSA is defined as the surface area over all polar atoms, primarily oxygen and nitrogen, including their attached hydrogens [66]. It is a key descriptor for predicting a molecule's ability to cross biological membranes, including the gastrointestinal barrier and the blood-brain barrier (BBB).
Hydrogen Bond Donors (HBD) and Acceptors (HBA)
HBD refers to the sum of all OH and NH groups, while HBA is the sum of all nitrogen and oxygen atoms [82]. These descriptors are critical for estimating a compound's capacity to form hydrogen bonds with biological targets and solvents, directly influencing solubility, permeability, and metabolism.
Molecular Weight (MW)
MW is the simplest descriptor but has profound implications. Increasing molecular weight can impact a compound's solubility, diffusion rate, and its ability to be absorbed via passive diffusion.
Table 1: Core Descriptor Definitions and Their Roles in ADMET
| Descriptor | Definition | Primary ADMET Influence |
|---|---|---|
| Lipophilicity (LogP/LogD) | Partition coefficient between n-octanol and water [66]. | Permeability, metabolic clearance, tissue distribution, toxicity risk [25]. |
| Topological Polar Surface Area (TPSA) | Surface area contributed by polar atoms (O, N, S, P and their attached H) [66]. | Membrane permeability, absorption, BBB penetration [66]. |
| Hydrogen Bond Donors (HBD) | Count of OH and NH groups [82]. | Solubility, permeability, and transporter efflux. |
| Molecular Weight (MW) | Mass of the molecule (in Daltons). | Diffusion rates, solubility, and passive permeation [82]. |
The Interplay of Descriptors in Governing ADMET Properties
The four descriptors function not independently, but as an interconnected system. Their collective values determine a molecule's position in physicochemical space, which in turn dictates its ADMET profile.
Integrated Impact on Absorption and Permeability
For a molecule to be effectively absorbed after oral administration, it must possess a balance between hydrophilicity (for dissolution) and lipophilicity (for membrane permeation). TPSA and HBD count are inversely correlated with passive permeability. For instance, a high TPSA (>140 Ų) generally indicates poor absorption, while a lower TPSA is favorable for intestinal permeability and BBB penetration [66]. Lipophilicity enhances permeability, but only up to a point; excessively high LogP can lead to poor solubility or sequestration in the membrane. The BOILED-Egg model is a proficient predictive tool that leverages both TPSA and LogP to simultaneously forecast gastrointestinal absorption (HIA) and brain access (BBB) [66].
Influence on Distribution and Metabolism
Lipophilicity is a key driver of a drug's distribution and its susceptibility to metabolism. Higher lipophilicity increases the volume of distribution and tissue binding but also accelerates metabolic clearance, primarily by hepatic CYP450 enzymes, which have a propensity to metabolize lipophilic compounds [25]. Molecules with lower LogP values (<1-2) are more likely to be cleared unchanged by the kidneys, leading to more linear pharmacokinetics [25]. Furthermore, increased lipophilicity and aromatic ring count are strongly correlated with higher risks of promiscuity, hERG channel inhibition, and general toxicity [82].
The following diagram illustrates the interconnected relationships between these core descriptors and key ADMET outcomes:
Quantitative Guidelines and Property-Based Design
Successful oral drugs typically occupy a well-defined region in physicochemical space. The Rule of Five (Ro5) established foundational guidelines, stating that poor absorption or permeation is more likely when a molecule violates two or more of the following: cLogP > 5, MW > 500, HBD > 5, HBA > 10 [82]. Subsequent research has refined these ranges for optimal drug-likeness.
Table 2: Optimal Ranges for Core Descriptors in Oral Drugs
| Descriptor | Optimal Range for Oral Drugs | Key Rationale |
|---|---|---|
| Lipophilicity (LogP/LogD) | 1 - 3 [25] | Balances permeability and solubility; minimizes metabolic clearance and toxicity risk. |
| TPSA | ~20 - 130 Ų | Favorable for passive permeability while maintaining sufficient solubility. |
| HBD | ≤ 5 [82] | Limits the number of H-bond donors, favoring membrane permeation. |
| MW | ≤ 500 [82] | Ensures manageable molecular size for adequate absorption and solubility. |
The Bioavailability Radar is a powerful visualization tool that encapsulates six key properties—lipophilicity, size, polarity, solubility, flexibility, and saturation—into a single, easy-to-interpret diagram. For a compound to be considered drug-like, its radar plot must fall entirely within the pink area defined for each axis [66]. This provides a rapid, holistic appraisal of a compound's drug-likeness, superior to checking individual parameters in isolation.
Computational and Experimental Methodologies
Integrated Workflow for ADMET Profiling
A modern drug discovery program leverages both computational and experimental techniques in an iterative cycle to optimize these descriptors. The following workflow outlines the key stages from initial design to experimental validation:
In Silico Prediction Protocols
Computational tools are indispensable for early-stage prediction. The following protocol can be applied to a library of compounds:
- Input Preparation: Draw 2D chemical structures or prepare a list of SMILES strings.
- Descriptor Calculation: Use a free web tool like SwissADME [66].
- The tool calculates key physicochemical properties (MW, TPSA, HBD, HBA).
- It provides a consensus LogP from five different predictive methods (iLOGP, XLOGP3, WLOGP, MLOGP, SILICOS-IT) for a robust estimate [66].
- Drug-Likeness Evaluation:
- Apply the Rule of Five filter.
- Generate and inspect the Bioavailability Radar plot.
- Use the BOILED-Egg model to predict passive absorption and brain penetration simultaneously [66].
- Analysis: Prioritize compounds whose predicted parameters fall within the optimal ranges described in Table 2.
Experimental Protocol for Lipophilicity Determination via RP-TLC
While computational predictions are fast, experimental validation is crucial. Reversed-Phase Thin Layer Chromatography (RP-TLC) is a robust, low-cost method for determining lipophilicity [15].
Table 3: Research Reagent Solutions for RP-TLC Lipophilicity Assay
| Reagent/Material | Function in the Protocol |
|---|---|
| RP-18 TLC Plates | Stationary phase; silica gel modified with octadecyl chains for a non-polar surface. |
| Acetone-TRIS Buffer (pH 7.4) | Mobile phase; a mixture of an organic modifier and aqueous buffer to simulate physiological pH. |
| Analytical Standard (Pure Compound) | The sample whose lipophilicity is being determined. |
| Developing Chamber | A sealed tank to contain the mobile phase atmosphere during chromatographic development. |
| Detection System (UV Lamp) | To visualize the separated compound spots if they are UV-active. |
Detailed Step-by-Step Protocol [15]:
- Plate Preparation: Cut the RP-18 TLC plate into small strips (e.g., 4 x 8 cm). Using a soft pencil, lightly draw a starting line about 1.5 cm from the bottom edge.
- Sample Application: Dissolve the test compound in a volatile solvent (e.g., methanol). Using a capillary tube, apply a small, concentrated spot of the solution onto the starting line. Allow the spot to dry completely.
- Mobile Phase Preparation: Prepare a series of mobile phases with varying ratios of acetone to TRIS buffer (e.g., 60:40, 70:30, 80:20 v/v). Pour approximately 20 mL of one mobile phase into the developing chamber, seal the lid, and allow it to saturate for 15-20 minutes.
- Chromatographic Development: Place the TLC strip vertically into the chamber, ensuring the starting line is above the mobile phase level. Replace the lid and allow the mobile phase to ascend until it is about 1-2 cm from the top of the plate.
- Detection and Measurement: Remove the plate from the chamber and mark the solvent front. Dry the plate and visualize the compound spot under a UV lamp (if applicable). Circle the spot and measure the distance from the start line to the center of the spot (distance traveled by solute, D_s). Also, measure the total distance traveled by the solvent front (D_f).
- Data Calculation: For each mobile phase composition, calculate the retardation factor, R_F = D_s / D_f. Then, compute the lipophilicity parameter R_M using the formula: R_M = log (1 / R_F - 1).
- Determination of RM0 (Extrapolated Lipophilicity): Plot the R_M values against the volume fraction of the organic modifier (e.g., acetone, φ) in the mobile phase. The y-intercept (R_M at φ = 0) of the resulting line is the RM0 value, which is considered the chromatographic descriptor of lipophilicity comparable to LogP [15].
Case Studies in Rational Drug Design
Optimizing Diquinothiazines for Anticancer Activity
A 2024 study on anticancer diquinothiazine hybrids exemplifies the practical integration of these principles. Researchers first determined the experimental lipophilicity (RM0) of 15 novel compounds using the RP-TLC protocol. They then compared these values with computationally derived LogP values from eight different algorithms (e.g., iLOGP, XLOGP3) to identify the most accurate predictor for their chemical series (iLOGP showed the biggest similarity). Finally, they used web tools like SwissADME and pkCSM to predict a wide range of ADMET parameters, correlating them with the measured lipophilicity to build a comprehensive pharmacokinetic profile early in the development process [15].
Leveraging Ligand Efficiency Metrics
To avoid the pitfalls of molecular inflation, modern medicinal chemistry employs efficiency metrics that normalize biological activity by molecular size or lipophilicity. Lipophilic Ligand Efficiency (LLE or LipE) is a critical metric, calculated as pIC50 (or pKi) - LogP (or LogD) [82]. This metric penalizes high lipophilicity, guiding chemists to seek potent compounds without resorting to excessive lipophilicity, thereby reducing the risk of poor solubility and promiscuous toxicity. For a candidate drug, an LLE > 5 is generally considered favorable, ensuring that potency is not achieved at the expense of a problematic ADMET profile [82].
The integration of lipophilicity with TPSA, HBD, and molecular weight is not merely a theoretical exercise but a practical necessity in contemporary drug discovery. These descriptors form a tightly interconnected network that dictates a molecule's fate in vivo. By applying the quantitative guidelines, utilizing the robust computational and experimental protocols outlined in this guide, and learning from successful case studies, researchers can systematically navigate the complex ADMET landscape. A deliberate, property-based design strategy that respects the balanced interplay of these core descriptors significantly enhances the probability of transforming a potent molecule into a safe and effective medicine.
Utilizing Lipinski's Rule of Five and Other Drug-Likeness Filters in Design
The high attrition rate of drug candidates in clinical trials, predominantly due to unfavorable pharmacokinetics or unacceptable toxicity, underscores the critical need for effective early-stage screening strategies [84]. The concept of "drug-likeness" provides a crucial framework for this screening, offering guidelines to select compounds with a higher probability of success. Among these guidelines, Lipinski's Rule of Five (Ro5) has emerged as a fundamental principle for predicting oral activity in humans [85]. This technical guide examines the application of Ro5 and subsequent drug-likeness filters within the broader context of a thesis investigating the profound impact of lipophilicity on Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties. For drug development professionals, understanding these filters is not merely an academic exercise but a practical necessity for prioritizing compounds with desirable bioavailability and safety profiles during the early phases of discovery [84].
Core Principles of Lipinski's Rule of Five
Formulated by Christopher A. Lipinski in 1997, the Rule of Five is a cornerstone of drug-likeness evaluation. It is predicated on the observation that most orally administered drugs are relatively small and moderately lipophilic molecules [85]. The rule states that an orally active drug is likely to have no more than one violation of the following criteria, all values of which are multiples of five, hence the name [85] [86]:
- Molecular weight (MW) ≤ 500 Daltons
- Number of Hydrogen Bond Donors (HBD) ≤ 5
- Number of Hydrogen Bond Acceptors (HBA) ≤ 10
- Calculated octanol-water partition coefficient (ClogP) ≤ 5
The partition coefficient (Log P) is a central parameter, defined as the ratio of the solubility of the un-ionized drug in 1-octanol to its solubility in water, serving as a key measure of lipophilicity [86]. The Rule of Five primarily describes molecular properties critical for a drug's pharmacokinetics in the human body, particularly its absorption and permeability, though it does not predict pharmacological activity [85]. Adherence to Ro5 has been correlated with lower attrition rates in clinical trials, thereby increasing a candidate's chance of reaching the market [85].
Limitations and the Evolution of Drug-Likeness Rules
Despite its widespread influence, the Rule of Five is not without limitations. Its primary assumption is that passive diffusion is the dominant mechanism for cellular entry, largely ignoring the role of transporters [85]. Critics note that only about 50% of orally administered new chemical entities actually obey it, and many natural products, such as macrolides and peptides, are effective despite violating its precepts [85]. Furthermore, an analysis of FDA-approved small-molecule protein kinase inhibitors reveals a trend toward higher molecular weights, with approximately 40% exceeding the 500 Da criterion [86].
To address these limitations and refine the prediction of drug-likeness, several extensions and variants of Ro5 have been developed, as summarized in Table 1.
Table 1: Key Drug-Likeness Rules and Filters Beyond the Rule of Five
| Rule/Filter | Key Criteria | Primary Objective |
|---|---|---|
| Ghose Filter [85] | - Log P between -0.4 to +5.6- Molar refractivity from 40 to 130- MW from 180 to 480- Number of atoms from 20 to 70 | Qualifying ranges for lipophilicity and size, setting lower limits. |
| Veber's Rule [85] | - ≤ 10 rotatable bonds- Polar Surface Area (TPSA) ≤ 140 Ų | Better discrimination of oral bioavailability, questioning the 500 MW cutoff. |
| Lead-like (Rule of Three) [85] | - Log P ≤ 3- MW < 300- HBD ≤ 3- HBA ≤ 3- Rotatable bonds ≤ 3 | To define "lead-like" compounds in screening libraries, allowing room for optimization while maintaining drug-likeness. |
| Fraction Lipophilicity Index (FLI) [87] | A composite metric combining Log P and Log D in a weighted manner. A drug-like FLI range of 0–8 was established, covering >90% of highly absorbed drugs. | Account for both intrinsic (Log P) and apparent (Log D) lipophilicity, providing a better assessment for ionizable compounds. |
Modern Methodologies: Integrating AI and Multidimensional Screening
The field has evolved from relying on simple rule-based filters to employing sophisticated, AI-powered platforms that enable a multidimensional evaluation of drug-likeness. These platforms assess critical dimensions beyond basic physicochemical properties, including toxicity, binding affinity, and synthesizability [88].
Comprehensive Evaluation with druglikeFilter
The druglikeFilter framework exemplifies this modern approach, using deep learning to evaluate compounds across four pillars [88]:
- Physicochemical Rule Evaluation: The tool calculates 15 common physicochemical properties (e.g., MW, HBD, HBA, ClogP, TPSA) and integrates 12 practical drug-likeness rules to filter out non-druggable and promiscuous compounds [88].
- Toxicity Alert Investigation: It screens for approximately 600 structural alerts associated with various toxicities (acute toxicity, skin sensitization, genotoxicity) and incorporates predictive models for specific endpoints like cardiotoxicity (hERG blockade) [88].
- Binding Affinity Measurement: A dual-path analysis is employed. For targets with known structures, molecular docking (using AutoDock Vina) is performed. For targets with only sequence information, a sequence-based AI model (transformerCPI2.0) predicts compound-protein interactions [88].
- Compound Synthesizability Assessment: The framework estimates synthetic accessibility and integrates Retro*, a neural-based retrosynthetic algorithm, to plan viable synthetic routes, addressing a key practical challenge in drug development [88].
The following workflow diagram illustrates the integrated process of this multidimensional screening approach.
Experimental Protocols for Key Assays
Measuring Lipophilicity
Accurate lipophilicity measurement is fundamental. While computational estimates are used initially, experimental validation is crucial.
- Protocol: High-Throughput 1-Octanol/Water Shake Flask for Compound Mixtures [89]
- Principle: This method measures the distribution of compounds between 1-octanol and aqueous phases, reflecting their intrinsic lipophilicity (Log P) or apparent lipophilicity at a specific pH (Log D).
- Procedure:
- Prepare a mixture of up to 10 compounds dissolved in a suitable solvent.
- Mix equal volumes of 1-octanol (pre-saturated with water) and aqueous buffer (e.g., pH 7.4 for Log D₇.₄ or pH 5.5 for acids, pre-saturated with 1-octanol).
- Add the compound mixture to the biphasic system and shake vigorously to allow partitioning.
- Centrifuge to separate the phases clearly.
- Analyze the concentration of each compound in both phases using High-Performance Liquid Chromatography coupled with Tandem Mass Spectrometry (LC/MS/MS).
- Calculation: The Log D is calculated as the logarithm of the ratio of the compound's concentration in the octanol phase to its concentration in the aqueous phase. This high-throughput adaptation allows for the simultaneous measurement of multiple compounds, making it suitable as a primary screen [89].
In Silico Binding Affinity Assessment (Docking)
- Protocol: Structure-Based Binding Affinity Prediction using AutoDock Vina [88]
- Principle: Molecular docking evaluates the binding pose and affinity of a ligand to a protein target based on geometric complementarity and force field calculations.
- Procedure:
- Protein Preparation: The uploaded 3D protein structure (e.g., from PDB) is preprocessed. This involves cleaning (removing water molecules, cofactors), reconstructing missing bonds, and adding hydrogen atoms.
- Ligand Preparation: Ligand structures are converted into 3D, and energy is minimized.
- Grid Box Definition: The binding pocket on the protein is defined based on the known location of a native ligand or through user-defined coordinates and dimensions.
- Docking Run: AutoDock Vina performs a search algorithm to find the optimal binding conformation and calculates a docking score (in kcal/mol) representing the estimated binding affinity.
- Post-processing: The compounds are ranked based on their docking scores, and the poses are analyzed for key interactions (e.g., hydrogen bonds, hydrophobic contacts).
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Research Reagent Solutions for Drug-Likeness and ADMET Studies
| Reagent / Resource | Function / Application | Key Features |
|---|---|---|
| 1-Octanol / Buffer Systems [89] | Experimental determination of partition (Log P) and distribution (Log D) coefficients via the shake-flask method. | Industry-standard for measuring lipophilicity; can be adapted for high-throughput using LC/MS/MS. |
| MedChem Designer [87] | Software for calculating molecular properties, including Log P, Log D, and the Fraction Lipophilicity Index (FLI). | Provides calculated properties for rapid in silico screening and profiling. |
| ClogP for Windows [87] | Software for calculating the partition coefficient for comparison with Lipinski's Ro5. | A widely used algorithm for Log P estimation. |
| RDKit [88] | An open-source cheminformatics toolkit used for calculating molecular descriptors and estimating synthetic accessibility. | Programmable via Python; integrates into automated screening workflows like druglikeFilter. |
| AutoDock Vina [88] | Open-source molecular docking software for structure-based prediction of binding affinity. | Fast, widely used; integrated into platforms for automated binding affinity measurement. |
| druglikeFilter Web Server [88] | A comprehensive deep learning-based web tool for multidimensional drug-likeness evaluation. | Integrates physicochemical, toxicity, binding, and synthesizability evaluation in one platform. |
| SwissADME Web Server [84] | A free web tool to evaluate pharmacokinetics, drug-likeness, and medicinal chemistry properties. | User-friendly interface for rapid predictive modeling of ADME-related parameters. |
Lipinski's Rule of Five remains an indispensable starting point for assessing drug-likeness, particularly for orally administered agents. However, its limitations have catalyzed the development of more nuanced filters and sophisticated AI-driven tools. The critical role of lipophilicity, as captured not only by Ro5 but also by advanced metrics like the Fraction Lipophilicity Index, is undeniable in its profound influence on ADMET outcomes [87]. The future of drug design lies in the integrated, multidimensional application of these tools. Platforms like druglikeFilter represent a paradigm shift, moving beyond simple rule-based filtering to a holistic evaluation that encompasses safety, efficacy, and practical synthesizability early in the discovery process [88]. For researchers, this integrated approach provides a more robust and reliable strategy for navigating the complex chemical space and prioritizing the most viable candidates for development, thereby increasing the efficiency and success rate of drug discovery.
The prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties represents a critical challenge in modern drug discovery. Among various molecular characteristics, lipophilicity serves as a master variable influencing a wide array of ADMET properties and overall druggability. Compounds with poor solubility and inappropriate lipophilicity face significantly higher attrition rates during development, contributing to staggering costs that often exceed $2 billion per approved drug [90]. Undesirable ADMET properties constitute a leading cause of clinical-phase failure, creating an urgent need for accurate early-stage prediction methods [90].
Graph Neural Networks (GNNs) have emerged as transformative tools for ADMET prediction by directly learning from molecular structures. Unlike traditional quantitative structure-activity relationship (QSAR) models that rely on pre-defined molecular descriptors, GNNs operate on natural graph representations of molecules, where atoms constitute nodes and bonds form edges [90] [91]. This approach bypasses the computationally expensive retrieval and selection of molecular descriptors while capturing subtle structure-property relationships that often elude descriptor-based methods [90] [92]. The integration of GNNs into ADMET prediction pipelines is particularly valuable for understanding and optimizing the complex effects of lipophilicity across the entire drug discovery workflow.
Molecular Graph Representation: From Structure to Computational Framework
Fundamentals of Molecular Graph Theory
The foundation of GNN-based ADMET prediction lies in the mathematically rigorous representation of molecules as graphs. Formally, a molecular graph is defined as G = (V, E), where V represents the set of nodes (atoms) and E represents the set of edges (bonds) connecting these nodes [90]. In computational implementations, this theoretical framework translates into concrete matrix representations:
- Adjacency Matrix (A): An N×N matrix where N is the number of atoms, with elements a_ij = 1 if atoms i and j are connected, and 0 otherwise [90]
- Node Feature Matrix (H): An N×D matrix containing D-dimensional feature vectors for each atom, encoding atomic properties [90]
For undirected molecular graphs (where bonds lack directionality), the adjacency matrix is symmetric, and self-connections are typically included by setting diagonal elements a_ii = 1 [90].
Atomic Feature Engineering
The node feature matrix incorporates essential chemical information that enables GNNs to learn meaningful structure-property relationships. Current implementations typically include the features detailed in Table 1, with each feature one-hot encoded and concatenated to form the complete atomic feature vector [90].
Table 1: Atomic Features for Node Representation in Molecular Graphs
| Atomic Feature | Possible Values | Chemical Significance |
|---|---|---|
| Atom Type | Atomic numbers 1-101 | Element identity and core properties |
| Formal Charge | -3, -2, -1, 0, 1, 2, 3, "Extreme" | Electrostatic interactions, ionization |
| Hybridization Type | S, SP, SP2, SP3, SP3D, SP3D2, "Other" | Molecular geometry and bond character |
| In a Ring | 0 (No), 1 (Yes) | Structural complexity and rigidity |
| In an Aromatic Ring | 0 (No), 1 (Yes) | π-electron systems and delocalization |
| Chirality | Unspecified, Clockwise, Counter-clockwise, Other | Stereochemistry and enantioselectivity |
Advanced Graph Representation Strategies
Sophisticated GNN architectures extend beyond simple graph representation by implementing specialized adjacency matrices for different bond types. As illustrated in Figure 1, researchers often construct five distinct adjacency matrices (A₁-A₅) for each molecule: one considering all bonds, and four others focusing exclusively on single, double, triple, and aromatic bonds, respectively [90]. This multi-faceted representation enables the model to capture nuanced electronic and structural features that directly influence molecular properties, including critical lipophilicity parameters.
Figure 1: Workflow for Molecular Graph Representation from SMILES Notation
GNN Architectures for ADMET Prediction
Core Architectural Frameworks
Multiple GNN architectures have been adapted and optimized for ADMET prediction tasks, each with distinct advantages for capturing molecular structure-property relationships:
- Message Passing Neural Networks (MPNNs): Iteratively update node representations by passing messages along edges, effectively capturing local chemical environments [93]
- Graph Attention Networks (GATs): Incorporate attention mechanisms to weigh the importance of neighboring nodes differently, mimicking chemical intuition about significant molecular regions [90]
- Graph Isomorphism Networks (GINs): Designed to be as powerful as the Weisfeiler-Lehman graph isomorphism test, addressing limitations of standard MPNNs in distinguishing certain molecular structures [93]
Composite and Specialized Architectures
Recent research has demonstrated that composite architectures combining the strengths of different GNN frameworks often achieve superior performance for specific ADMET tasks. The Directed Edge Graph Isomorphism Network (D-GIN) exemplifies this trend by integrating the directed-edge message passing of D-MPNN with the structural discrimination power of GIN [93]. This architecture has shown particular effectiveness for predicting lipophilicity parameters (logD/logP) and aqueous solubility (logS), achieving state-of-the-art performance with R² values exceeding 0.909 on benchmark datasets [93] [92].
For cytochrome P450 inhibition prediction—critical for metabolism assessment—attention-based GNNs have demonstrated exceptional capability by focusing computational resources on molecular substructures most relevant to enzyme binding [90] [91]. These models process information through a bottom-up approach, from local atomic environments to global molecular features, effectively capturing both substrate and inhibitor characteristics for major CYP isoforms including CYP3A4, CYP2D6, and CYP2C9 [90] [91].
Experimental Protocols and Methodologies
Data Preparation and Curation
Robust ADMET prediction begins with rigorous data curation. For lipophilicity prediction, experimental protocols typically utilize the Delaney lipophilicity dataset containing experimentally derived logD and logP values at pH 7.4, often complemented by aqueous solubility data [93]. Standard preprocessing includes:
- Data Neutralization: Adjusting molecular representations to neutral forms
- Salt Stripping: Removing counterions and salt forms to isolate the core molecular structure
- Range Filtering: Eliminating extreme outliers (e.g., logS values < -10.0 or > 0.0) that may represent measurement artifacts [93]
Datasets are typically partitioned into training, validation, and test sets using an 81:9:10 ratio, with consistent random seeds to ensure reproducible comparisons across model architectures [93].
Training Strategies and Optimization
Advanced training methodologies have been developed specifically to address data limitations in ADMET prediction:
- Multi-Task Learning (MTL): Simultaneously trains on multiple ADMET endpoints, allowing information sharing across related properties and effectively increasing usable samples [94] [95]
- Transfer Learning (PCFE): Pretrains GNNs on large-scale computational data (low-fidelity) followed by fine-tuning on smaller experimental datasets (high-fidelity), dramatically improving performance for data-scarce properties like log D₇.₄ [92]
- Adaptive Auxiliary Task Selection: Employs status theory and maximum flow algorithms to intelligently select which auxiliary tasks will most benefit primary task learning [94]
Table 2: Performance Comparison of GNN Architectures on Key ADMET Properties
| GNN Architecture | ADMET Property | Performance Metric | Value | Reference |
|---|---|---|---|---|
| cx-Attentive FP | log D₇.₄ | R²test | 0.909 | [92] |
| D-GIN | Lipophilicity (logP) | RMSE | Not specified | [93] |
| Attention-based GNN | CYP450 Inhibition | AUC | 0.87 | [90] |
| MTGL-ADMET | Multiple endpoints | Outperformed STL/MTL baselines | Significant improvement | [94] |
| GNNMT+FT | 7 of 10 ADME parameters | Highest performance | Superior to conventional methods | [95] |
Explainability and Interpretation Methods
Model interpretability is crucial for translating predictions into actionable chemical insights during lead optimization. Integrated Gradients (IG) has emerged as a powerful method for quantifying feature contributions, enabling both visual and quantitative identification of atoms and substructures influencing specific ADMET predictions [95]. Complemented by attention mechanisms that highlight chemically significant molecular regions and SHAP (Shapley Additive Explanations) analysis for descriptor importance, these methods provide transparent windows into model decision-making [92] [95]. Validation studies demonstrate strong alignment between computational attention patterns and established chemical knowledge, particularly for lipophilicity-modifying substituents like hydrocarbon groups, halogens, and polar functional groups [92] [95].
Table 3: Key Research Reagents and Computational Resources
| Resource | Type | Function | Access |
|---|---|---|---|
| pkCSM | Web Server | Predicts pharmacokinetic properties using graph-based signatures | Freely available for academic use [68] |
| ChemLogD | Web Server | Specialized log D₇.₄ prediction using cx-Attentive FP model | http://tools.scbdd.com/chemlogd/ [92] |
| TDC (Therapeutics Data Commons) | Platform | Benchmarking and evaluation framework for ADMET prediction models | Open access [90] |
| DruMAP | Database | Curated ADME experimental data for model training | Publicly available from NIBIOHN [95] |
| GraphSAGE | Framework | GNN implementation optimized for scalability to large molecular sets | Open source [96] |
| kMoL | Software Package | GNN construction and implementation toolkit | Research use [95] |
Integrated Workflow: From Molecular Design to ADMET Optimization
The complete GNN-driven ADMET prediction pipeline integrates multiple components into a cohesive workflow that directly addresses lipophilicity challenges in drug development. As visualized in Figure 2, this process begins with molecular graph representation, proceeds through specialized GNN architectures trained via advanced learning strategies, and culminates in interpretable predictions that guide molecular design.
Figure 2: Integrated Workflow for GNN-Driven ADMET Prediction and Optimization
This end-to-end framework enables researchers to rapidly cycle through design-prediction-optimization iterations, systematically addressing lipophilicity and other critical ADMET parameters early in the discovery process. By highlighting specific structural features contributing to undesirable properties, the approach moves beyond black-box prediction to provide chemically intuitive guidance for molecular modification [95].
Graph Neural Networks have fundamentally transformed ADMET prediction capabilities, particularly for understanding and optimizing the complex role of lipophilicity in drug development. By directly learning from molecular structures and leveraging advanced training paradigms like multi-task learning and transfer learning, GNNs achieve state-of-the-art performance across diverse ADMET endpoints while providing interpretable insights that guide lead optimization.
Future advancements will likely focus on several key areas: improving generalization to novel chemical spaces, integrating real-time experimental validation, developing more sophisticated explainability frameworks, and creating unified platforms that encompass the entire drug discovery pipeline from target identification to toxicity assessment [91]. As these computational approaches continue to mature, they promise to further reduce late-stage attrition rates and accelerate the delivery of safer, more effective therapeutics.
Troubleshooting ADMET Issues and Strategies for Lipophilicity Optimization
Identifying and Correcting Poor Solubility in Highly Lipophilic Compounds
Lipophilicity, most commonly measured by the octanol/water partition coefficient (LogP), is a fundamental physicochemical property that plays a critical role in drug discovery and development. As a key descriptor, it profoundly influences a compound's Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profile [97] [6]. Highly lipophilic compounds face significant pharmaceutical challenges, particularly poor aqueous solubility, which directly impacts drug bioavailability [36]. Nearly 90% of drug candidates in development pipelines are poorly water-soluble, with a substantial portion belonging to Biopharmaceutics Classification System (BCS) classes II or IV [98] [99]. This review examines the intricate relationship between lipophilicity and solubility, presents advanced formulation strategies to overcome these challenges, and discusses analytical and computational frameworks for optimizing drug candidates within the context of comprehensive ADMET profiling.
The pursuit of potent compounds often leads medicinal chemists toward higher molecular weight and lipophilicity, creating molecules with increasingly complex structures [36]. This "molecular obesity" trend presents significant challenges in later development stages, as highly lipophilic compounds often demonstrate poor aqueous solubility, increased metabolic clearance, promiscuous binding leading to off-target effects, and ultimately, higher clinical attrition rates [97]. Maintaining lipophilicity within an optimal range (typically LogP < 5) through monitoring of lipophilic efficiency indices such as LLE (Lipophilic Ligand Efficiency) and LELP (Ligand Efficiency-dependent Lipophilicity) can significantly improve compound quality and the likelihood of therapeutic success [97].
The Lipophilicity-Solubility Relationship: Fundamental Principles
Molecular Determinants of Solubility
According to the General Solubility Equation (GSE) for organic nonelectrolytes, two key factors primarily influence a substance's solubility: the melting point (Tm) and the octanol-water partition coefficient (LogP) [98]. These parameters allow for a practical classification of poorly water-soluble drugs into two categories:
- 'Brick-dust' molecules: Characterized by high melting points due to strong crystal lattice energies, leading to limited solubility despite potentially moderate lipophilicity.
- 'Grease-ball' molecules: Exhibiting high lipophilicity (LogP) as the primary limiting factor for solubility, often with low melting points [98].
This distinction is crucial for selecting appropriate formulation strategies, as each category responds differently to various solubilization techniques. For highly lipophilic compounds, the 'grease-ball' classification typically applies, where solvation—rather than solid-state properties—poses the primary limitation to dissolution [98].
Table 1: Classification of Poorly Water-Soluble Drugs Based on Solubility-Limiting Factors
| Category | Primary Limiting Factor | Key Characteristics | Typical Formulation Approach |
|---|---|---|---|
| Brick-dust Molecules | High melting point (solid-state properties) | Strong crystal lattice energy, high melting point | Modification of solid state (e.g., solid dispersions, drug nanoparticles) |
| Grease-ball Molecules | High lipophilicity (LogP) | Limited solvation, low melting point | Lipid-based formulations, emulsion systems |
Lipophilicity and ADMET Interrelationships
Lipophilicity influences nearly all aspects of a drug's pharmacokinetic profile. Highly lipophilic compounds (LogP > 5) face an increased risk of poor solubility, rapid metabolism, promiscuous binding to off-target proteins, and toxicity concerns [97] [6]. Conversely, compounds with insufficient lipophilicity may demonstrate inadequate membrane permeability and poor target engagement. This delicate balance underscores the importance of maintaining lipophilicity within an optimal range throughout drug optimization campaigns.
The ionization state (pKa) of a drug further modulates the interplay between lipophilicity and solubility, as it directly influences the compound's charge state across physiological pH gradients—from the highly acidic environment of the stomach (pH ~2) to the neutral intracellular milieu (pH ~7.5) [6]. For ionizable compounds, the pH-partition hypothesis describes how distribution coefficients (LogD) vary with pH, significantly impacting gastrointestinal absorption and tissue distribution patterns.
Formulation Strategies for Lipophilic Compounds
Lipid-Based Formulation Systems
Lipid-based formulations represent a primary strategy for enhancing the bioavailability of 'grease-ball' molecules with high lipophilicity [98]. These systems work by maintaining the drug in a solubilized state throughout the gastrointestinal transit, facilitating absorption via lymphatic transport, and reducing first-pass metabolism. The approaches include:
- Self-Emulsifying Drug Delivery Systems (SEDDS): isotropic mixtures of oils, surfactants, and co-solvents that form fine oil-in-water emulsions upon mild agitation in aqueous media.
- Lipid Solutions: non-polar formulations where the drug is dissolved in digestible oils such as medium-chain triglycerides.
- Lipid Suspensions: systems where the drug is suspended in a lipid base, with dissolution occurring during lipid digestion.
The selection of lipid excipients depends on the drug's solubility in various lipids, the required drug loading, and the intended release profile. Long-chain triglycerides enhance lymphatic transport, while medium-chain triglycerides often provide superior solvation capacity for highly lipophilic compounds.
Drug Nanoparticle Technologies
Nanoparticle engineering significantly increases the surface area-to-volume ratio of drug particles, thereby enhancing dissolution rates according to the Noyes-Whitney and Nernst-Brunner equations [98]. For highly lipophilic compounds, targeting particle sizes below approximately 300 nm can dramatically improve bioavailability [98]. Two primary approaches dominate this field:
- Top-Down Methods: Nanomilling techniques use mechanical energy to reduce particle size. Wet media milling employs grinding beads to break down drug particles in a liquid suspension, achieving sizes below 200 nm typically within 60-120 minutes [98]. Process parameters must be optimized to minimize contamination from bead wear and prevent Ostwald ripening—a phenomenon where smaller particles dissolve and re-deposit onto larger crystals due to their higher solubility [98].
- Bottom-Up Methods: Precipitation techniques involve dissolving the drug in a solvent and then adding this solution to an anti-solvent, causing spontaneous nucleation and formation of nanoscale particles. This approach avoids mechanical stress but requires careful control of supersaturation to ensure consistent particle size distribution.
Stabilizing nanoparticles against agglomeration is critical and achieved through electrostatic stabilization (charged surfactants), steric stabilization (non-ionic polymers), or electrosteric stabilization (combination of both) [98]. Hansen Solubility Parameters (HSP) have emerged as a valuable tool for rationally selecting optimal stabilizers rather than relying solely on empirical screening [98].
Solid Dispersion Systems
Solid dispersions incorporate the drug in a hydrophilic carrier matrix at the molecular or micro-particulate level, creating high-energy amorphous states with enhanced dissolution properties. For highly lipophilic compounds, this approach can overcome the solvation limitations by creating a more favorable microenvironment for water penetration and drug release. Common carrier systems include:
- Polymer-based systems: Using polymers such as polyvinylpyrrolidone (PVP), hydroxypropyl methylcellulose (HPMC), or copolymers like polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer (Soluplus).
- Mesoporous silica carriers: Inorganic matrices with high surface area and tunable pore sizes that confine drug molecules in amorphous states.
The selection of appropriate carriers depends on drug-polymer miscibility, stability considerations, and manufacturing feasibility. Recent advances focus on achieving and maintaining supersaturation after dissolution to maximize intestinal absorption.
Table 2: Comparison of Major Formulation Strategies for Highly Lipophilic Compounds
| Strategy | Mechanism of Action | Best Suited For | Key Challenges |
|---|---|---|---|
| Lipid-Based Formulations | Maintains drug in solubilized state, enhances lymphatic transport | Grease-ball molecules with high LogP | Drug loading limitations, stability of lipid excipients |
| Drug Nanoparticles | Increases surface area for enhanced dissolution rate | Both brick-dust and grease-ball molecules | Physical stability, Ostwald ripening, contamination from milling |
| Solid Dispersions | Creates high-energy amorphous states in hydrophilic matrix | Compounds with some polar functionality | Physical stability, crystallization tendency, scale-up |
Experimental Protocols and Methodologies
Nanoparticle Preparation via Wet Media Milling
Objective: To produce stable drug nanoparticles with enhanced dissolution properties for highly lipophilic compounds.
Materials:
- Poorly water-soluble drug compound
- Stabilizers (e.g., Poloxamer 407, HPMC, PVP, sodium lauryl sulfate)
- Deionized water
- Grinding beads (yttrium-stabilized zirconium oxide, 0.3-0.5 mm diameter)
- Planetary ball mill or stirred media mill
Procedure:
- Prepare a suspension containing 10-40% (w/w) drug in aqueous stabilizer solution.
- Add grinding beads to the milling chamber at a filling ratio of 30-50% of chamber volume.
- Load the drug suspension into the milling chamber.
- Operate the mill at optimal parameters (typically 60-120 minutes) with active cooling to maintain temperature below 40°C.
- Separate the nanoparticles from grinding beads using a mesh sieve.
- Characterize the resulting nanosuspension for particle size (dynamic light scattering), morphology (SEM), crystalline form (PXRD), and dissolution profile [98].
Critical Considerations:
- Stabilizer selection and concentration must be optimized to prevent agglomeration through steric and/or electrostatic stabilization.
- Process parameters (bead size, milling time, energy input) significantly impact final particle size distribution and potential contamination from bead wear.
- Long-term physical stability must be assessed, including potential for Ostwald ripening and crystal growth during storage [98].
Solid Dispersion Preparation via Solvent Evaporation
Objective: To create a molecularly dispersed drug-polymer system with enhanced dissolution properties.
Materials:
- Poorly water-soluble drug
- Hydrophilic polymer carrier (e.g., PVP VA64, HPMCAS, Soluplus)
- Organic solvent (e.g., dichloromethane, methanol, acetone)
- Rotary evaporator or spray dryer
Procedure:
- Dissolve drug and polymer in suitable organic solvent at specific drug:polymer ratios (typically 1:1 to 1:4).
- For rotary evaporation: Transfer solution to round-bottom flask and evaporate under reduced pressure at controlled temperature (typically 40-60°C).
- For spray drying: Atomize the solution through a nozzle into a heated chamber to rapidly evaporate the solvent.
- Collect the resulting solid dispersion and dry under vacuum to remove residual solvent.
- Characterize the solid dispersion for solid state (PXRD, DSC), drug-polymer interactions (FTIR), dissolution performance, and physical stability under accelerated conditions.
Critical Considerations:
- Solvent selection must ensure complete dissolution of both drug and polymer.
- The manufacturing process must achieve complete solvent removal to meet safety specifications.
- Physical stability studies should monitor for crystallization over time under various temperature and humidity conditions.
Computational and Analytical Tools for Solubility Assessment
In Silico Prediction of Physicochemical Properties
Computational approaches enable early assessment of solubility challenges during drug design. Key predictive models include:
- Lipophilicity prediction: Calculated LogP (cLogP) models using fragmental or atom-based approaches provide estimates of partition coefficients [6].
- Solubility prediction: Quantitative Structure-Property Relationship (QSPR) models correlate molecular descriptors with thermodynamic solubility [6].
- ADMET profiling: Comprehensive tools like admetSAR 2.0 predict multiple ADMET endpoints simultaneously, providing a holistic assessment of drug-likeness [100].
Machine learning algorithms have significantly improved the accuracy of these predictions. Random Forest and gradient boosting methods often outperform traditional regression models, especially when trained on large, high-quality datasets [101]. The ADMET-score represents an advanced integrative approach, combining predictions from 18 different ADMET endpoints into a single composite score that evaluates overall drug-likeness [100].
Table 3: Computational Tools for Predicting Key Properties Relevant to Lipophilic Compounds
| Property | Prediction Method | Typical Accuracy | Application in Design |
|---|---|---|---|
| Lipophilicity (LogP) | Fragmental/atom-based methods, machine learning | RMSE ~0.5 log units [6] | Optimizing compound series toward optimal range (LogP 1-5) |
| Solubility (LogS) | QSPR models, group contribution methods | ~75% predictions within 1 logS unit [6] | Identifying potential solubility limitations early |
| ADMET-score | Ensemble of 18 binary classification models | Significantly discriminates approved vs. withdrawn drugs [100] | Comprehensive drug-likeness assessment |
Advanced Analytical Characterization
Chromatography coupled with mass spectrometry (MS) has emerged as a cornerstone technique for characterizing lipophilic compounds and their behavior in biological systems [102]. Key applications include:
- UHPLC-MS: Provides high-resolution separation of complex mixtures, particularly advantageous for nonpolar lipid molecules and lipophilic drug formulations [102].
- Mass spectrometry imaging: Enables spatial localization of drugs and metabolites in tissues, providing critical distribution data for highly lipophilic compounds.
- Stability assessment: Monitors chemical degradation and physical form changes in formulated products.
These analytical techniques provide essential data for understanding the in vivo performance of formulated lipophilic compounds, enabling rational formulation optimization based on mechanistic insights rather than empirical observation.
Visualizing Experimental Workflows and Strategic Approaches
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagents and Materials for Solubility Enhancement Studies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Yttrium-stabilized Zirconium Oxide Beads | Grinding media for nanomilling | Particle size reduction in wet media milling (0.3-0.5 mm diameter) [98] |
| Poloxamer 407 | Non-ionic polymeric stabilizer | Steric stabilization of drug nanoparticles [98] |
| Hydroxypropyl Methylcellulose (HPMC) | Matrix former, stabilizer | Solid dispersions, nanoparticle stabilization [98] |
| Medium-Chain Triglycerides | Lipid vehicle | Lipid-based formulations for grease-ball molecules [98] |
| Polyvinylpyrrolidone-vinyl Acetate (PVP VA64) | Amorphization polymer | Solid dispersion carrier for solubility enhancement |
| Sodium Lauryl Sulfate | Ionic surfactant | Electrostatic stabilization of nanosuspensions [98] |
| Mesoporous Silica Carriers | Inorganic carrier matrix | Adsorption and stabilization of amorphous drug [99] |
The challenge of poor solubility in highly lipophilic compounds remains a significant hurdle in drug development, yet substantial progress continues through advanced formulation technologies and strategic design approaches. The most successful outcomes emerge from integrated strategies that combine careful compound design with appropriate formulation selection based on the specific physicochemical properties of each molecule. Emerging trends including high-throughput analytical techniques, machine learning-guided formulation development, and continuous manufacturing processes promise to further enhance our ability to advance lipophilic drug candidates. As the field evolves, maintaining focus on the fundamental relationships between lipophilicity, solubility, and overall ADMET profile will continue to be essential for developing successful therapeutic agents from highly lipophilic compounds.
Mitigating Toxicity and Promiscuity Linked to Excessive Lipophilicity
Excessive lipophilicity in drug candidates is a primary contributor to high attrition rates in drug development, directly linked to poor solubility, increased metabolic clearance, promiscuous off-target binding, and elevated toxicity risks. This whitepaper examines the integral relationship between lipophilicity and ADMET properties, synthesizing current research findings to present a comprehensive framework for mitigation strategies. We provide quantitative data analysis, detailed experimental methodologies for lipophilicity assessment, and visualization of strategic workflows to guide researchers in optimizing compound profiles. The evidence demonstrates that systematic management of lipophilicity, particularly through metabolic soft-spot resolution rather than global lipophilicity reduction, significantly enhances the likelihood of clinical success while minimizing toxicity and promiscuity-related liabilities.
Lipophilicity, quantified as the partition coefficient (LogP) or distribution coefficient (LogD), represents a critical physicochemical parameter in drug design that profoundly influences pharmacokinetic and pharmacodynamic profiles. The consensus across numerous studies confirms that excessive lipophilicity correlates strongly with undesirable ADMET properties, including heightened risk of promiscuous target interactions, toxicity, rapid metabolic clearance, and poor aqueous solubility [103] [104]. Research analyzing over 500,000 compounds in the ChEMBL database reveals that the bias toward high in vitro potency often introduces suboptimal physicochemical properties, creating diametrically opposed challenges between achieving target potency and maintaining favorable ADMET characteristics [104]. This inherent conflict underscores the necessity for balanced approaches to lipophilicity management throughout drug discovery campaigns.
The fundamental mechanisms linking lipophilicity to adverse outcomes stem from basic chemical principles. Highly lipophilic compounds exhibit greater membrane permeability but reduced aqueous solubility, creating formulation challenges and limiting gastrointestinal absorption [14]. Furthermore, increased lipophilicity enhances nonspecific binding to phospholipid membranes, proteins, and off-target receptors, leading to promiscuous behavior and toxicity concerns such as hERG channel inhibition [103] [104]. Contemporary drug discovery must therefore navigate the delicate balance where sufficient lipophilicity enables target engagement while avoiding the ADMET liabilities associated with excessive hydrophobicity.
Quantitative Relationships: Lipophilicity and ADMET Liabilities
Key ADMET Impacts of Excessive Lipophilicity
Table 1: ADMET Liabilities Associated with Increasing Lipophilicity
| Lipophilicity Range (LogD₇.₄) | Primary ADMET Liabilities | Impact on Drug Discovery |
|---|---|---|
| <0 | Poor membrane permeability, limited absorption | Requires active transport mechanisms for adequate bioavailability |
| 0-3 | Optimal balance of solubility and permeability | Ideal range for most oral drugs with minimal liabilities |
| >3 | Increased metabolic clearance, promiscuous target binding | Higher risk of drug-drug interactions and toxicity |
| >4 | Significant solubility limitations, high plasma protein binding, increased hERG inhibition | Substantial attrition risk due to safety concerns and poor pharmacokinetics |
Analysis of Genentech's internal rat pharmacokinetic data encompassing 4,767 neutral small molecules demonstrates that both unsteady-state volume of distribution (Vdˢˢ,ᵘ) and unbound clearance (CLᵘ) tend to increase with lipophilicity, creating a complex interplay that often negates half-life extension attempts through simple lipophilicity reduction [103]. This dataset reveals that decreasing lipophilicity without addressing specific metabolic soft-spots typically reduces both clearance and volume of distribution simultaneously, resulting in negligible half-life improvement [103]. The matched molecular pair analysis from this study further indicates that transformations which improve metabolic stability without decreasing lipophilicity have an 82% probability of prolonging half-life, compared to only 30% for strategies focused solely on lipophilicity reduction [103].
Lipophilicity-Toxicity Relationships
Table 2: Toxicity Risks Associated with Elevated Lipophilicity
| Toxicity Endpoint | Relationship with Lipophilicity | Structural Modifications for Mitigation |
|---|---|---|
| hERG Inhibition | Strong positive correlation (p<0.001) | Reduce aromatic rings, introduce polar groups, decrease overall hydrophobicity |
| Mitochondrial Toxicity | Increased risk with LogP >3.5 | Incorporate carboxylic acids, reduce overall charge, improve structural rigidity |
| CYP450 Inhibition | Enhanced time-dependent inhibition with higher LogP | Remove metabolically labile groups, reduce lipophilicity near metabolic hot spots |
| Phospholipidosis | Significant correlation with ClogP >3 and presence of basic amines | Reduce hydrophobic chain length, modify basic pKa, introduce steric hindrance |
Beyond these specific toxicities, promiscuous target binding represents a critical concern with lipophilic compounds. Research indicates that oral drugs themselves frequently exhibit substantial off-target activity, with lipophilicity being a key determinant of promiscuity [104]. This off-target engagement potentially underlies idiosyncratic adverse drug reactions and remains a fundamental challenge in lead optimization phases. The data further suggests that oral drugs seldom possess nanomolar potency (averaging approximately 50 nM), contradicting the common screening cascade approach that prioritizes high in vitro potency as an initial filter [104].
Experimental Methodologies for Lipophilicity Assessment
Chromatographic Determination of Lipophilicity
Chromatographic techniques provide robust, high-throughput alternatives to traditional shake-flask methods for lipophilicity assessment. The reversed-phase thin-layer chromatography (RP-TLC) method offers particular advantages for rapid screening during early discovery stages:
Protocol: RP-TLC Lipophilicity Determination (RM₀ Value)
- Stationary Phase: Employ RP-18 silica plates with controlled carbon loading
- Mobile Phase: Prepare binary solvent systems with acetone and TRIS buffer (pH 7.4) in varying ratios
- Sample Application: Spot 0.1-1.0 μL of compound solution (1 mg/mL in methanol) in triplicate
- Chromatography Development: Develop chambers to a distance of 8-9 cm under saturated conditions
- Detection: Visualize under UV light (254 nm and 365 nm) or using appropriate chemical staining
- RM Value Calculation: Calculate RM = log(1/Rf - 1) for each mobile phase composition
- Lipophilicity Index: Extrapolate RM₀ value (RM value at 0% organic modifier) via linear regression [15]
The Organization for Economic Co-operation and Development (OECD) endorses reversed-phase high-performance liquid chromatography (RP-HPLC) as a preferred method for LogP determination, particularly for compounds challenging to assess via traditional shake-flask techniques [14]. HPLC offers superior efficiency, consistency, and applicability across diverse compound classes, including poorly water-soluble or volatile substances. When implementing HPLC methods, careful selection of stationary phase chemistry is crucial, with C18 columns serving as the benchmark for lipophilicity assessment. The linear-solvent-strength model is frequently employed in retention modeling, with the volume fraction of the organic modifier (φ) representing the most critical factor in retention modeling [14].
Computational Prediction Approaches
In silico tools provide rapid lipophilicity estimation during virtual compound screening, with multiple algorithms available:
Protocol: Computational Lipophilicity Prediction
- Input Preparation: Generate standardized molecular structures (canonical SMILES) using tools like Open Babel
- Algorithm Selection: Employ multiple prediction methods to assess consensus values:
- iLOGP: Incorporates implicit solvation models and quantum mechanical descriptors
- XLOGP3: Uses atom-based fragmentation with correction factors
- MLOGP: Implements topological descriptors and multivariate analysis
- Descriptor Calculation: Compute fundamental molecular properties (molecular weight, polar surface area, hydrogen bond donors/acceptors) using RDKit or similar toolkits
- Validation: Compare computational predictions against experimental chromatographic parameters (RM₀) for benchmark compounds [15] [6]
Studies comparing computational predictions with experimental RP-TLC data demonstrate that algorithms such as iLOGP show the strongest correlation with chromatographic measurements for certain chemical series [15]. Commercial platforms like Chemaxon's logP model have demonstrated superior performance in blind challenges, achieving the lowest root mean square error (RMSE) and highest R² values compared to other prediction tools [6]. For comprehensive ADMET assessment, integrated platforms like admetSAR3.0 provide prediction for 119 endpoints, incorporating both search and optimization modules to guide structural modification [105].
Strategic Framework for Mitigation
Matched Molecular Pair Transformations
Analysis of successful half-life extending transformations reveals that addressing metabolic soft-spots rather than global lipophilicity reduction produces more favorable outcomes. The most effective transformations include:
- Introduction of halogens (e.g., H→F, CH₃→F) to block metabolic sites while potentially increasing lipophilicity but decreasing clearance
- Bioisosteric replacement of metabolically labile groups (e.g., methyl→trifluoromethyl) despite minimal lipophilicity change
- Steric shielding of vulnerable sites through adjacent substitution to hinder enzymatic access without substantially altering overall hydrophobicity [103]
These strategies demonstrate that metabolic stability improvements can be achieved independently of lipophilicity reduction, contradicting conventional approaches that prioritize cLogP reduction as a primary optimization tactic.
Machine Learning-Enhanced Optimization
Contemporary ADMET optimization leverages machine learning (ML) models trained on comprehensive datasets to predict property endpoints. The integration of graph neural networks (GNNs) represents a significant advancement, as these models process molecular graph representations directly from SMILES notation without requiring precomputed molecular descriptors [106]. This bottom-up approach enables more accurate modeling of complex structure-property relationships, particularly for ADMET endpoints with multifactorial determinants.
Benchmark studies indicate that random forest models frequently outperform other algorithms for specific ADMET prediction tasks, though optimal model selection remains dataset-dependent [101]. Feature representation critically impacts model performance, with systematic feature selection proving more effective than concatenating multiple representations without justification [101]. For lipophilicity-dependent toxicity endpoints, models incorporating molecular descriptors alongside structural fingerprints demonstrate superior predictive accuracy for promiscuity and specific toxicity risks like hERG inhibition.
Table 3: Key Research Reagent Solutions for Lipophilicity and ADMET Research
| Tool/Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Chromatographic Systems | RP-TLC (C18 plates), RP-HPLC (C18 columns), HILIC columns | Experimental lipophilicity determination, high-throughput screening | Physicochemical profiling during lead optimization |
| Computational Platforms | admetSAR3.0, SwissADME, pkCSM, Chemaxon Calculators | In silico prediction of ADMET endpoints and physicochemical properties | Virtual screening, compound prioritization, design of novel analogs |
| Machine Learning Frameworks | RDKit, PyTorch, DGL-LifeSci, Chemprop | Molecular descriptor calculation, feature engineering, model development | Building customized prediction models for specific chemical series |
| Public Databases | ChEMBL, PharmaBench, TDC, PubChem | Source of curated ADMET data for model training and validation | Benchmarking, model development, read-across analyses |
| Specialized Software | KNIME, DataWarrior, PipelinePilot | Data preprocessing, visualization, and analysis workflow management | Processing high-throughput screening data, structural analytics |
The development of comprehensive benchmarking datasets like PharmaBench, which incorporates 52,482 entries from 14,401 bioassays processed through a multi-agent large language model system, addresses previous limitations in public ADMET data quality and relevance [20]. This advancement, coupled with tools like admetSAR3.0's optimization module (ADMETopt2) that employs matched molecular pair analysis to suggest structural modifications, provides researchers with increasingly sophisticated resources for navigating lipophilicity-optimization challenges [105].
Strategic management of lipophilicity represents a critical success factor in modern drug discovery, directly impacting toxicity, promiscuity, and overall compound developability. The evidence conclusively demonstrates that reflexive reduction of lipophilicity without addressing specific metabolic vulnerabilities frequently fails to improve key pharmacokinetic parameters like half-life. Instead, successful optimization requires targeted interventions informed by comprehensive ADMET profiling and computational guidance. Emerging capabilities in machine learning, expansive benchmarking datasets, and sophisticated predictive platforms now enable more nuanced approaches to balancing lipophilicity with other molecular properties. By adopting the integrated strategic framework and methodological tools outlined in this whitepaper, researchers can systematically mitigate lipophilicity-related liabilities while maintaining desired target engagement, ultimately enhancing the efficiency of drug discovery and reducing late-stage attrition.
The Blood-Brain Barrier (BBB) represents one of the most significant challenges in central nervous system (CNS) drug development. This highly selective cellular barrier lines the cerebral microvasculature, protecting the brain from xenobiotics while simultaneously restricting access for more than 98% of small-molecule drugs and nearly 100% of large-molecule therapeutics [107] [34] [108]. The BBB exists as a dynamic interface composed of specialized endothelial cells connected by tight junctions, surrounded by pericytes, astrocytes, and microglia that collectively maintain barrier integrity and function [34] [108]. This complex cellular network strictly controls the passage of substances from systemic circulation to the brain parenchyma through multiple transport mechanisms, including transmembrane diffusion, carrier-mediated transport, receptor-mediated transcytosis, and adsorptive-mediated transcytosis [107] [34]. Understanding these pathways and the properties that govern molecular transit across the BBB is fundamental to CNS drug optimization. This technical guide examines core strategies for enhancing brain penetration, with particular emphasis on the critical relationship between lipophilicity and ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties that determines therapeutic success in neurological disorders.
Blood-Brain Barrier Structure and Transport Mechanisms
Anatomical Composition
The BBB derives its restrictive properties from unique anatomical features that distinguish cerebral endothelial cells from peripheral vasculature. Brain endothelial cells form continuous non-fenestrated walls joined by elaborate tight junctions comprising junctional adhesion molecules, occludin, and claudin proteins [107] [34]. These junctions virtually eliminate paracellular transport between cells, while significantly reduced pinocytosis limits transcellular bulk flow [109] [108]. Additional contributions from astrocytes, pericytes, and the basal lamina create a neurovascular unit that collectively maintains CNS homeostasis [34] [108]. Pericytes cover approximately 100% of the CNS endothelium and play crucial roles in regulating tight junction formation, vascular development, and neuroinflammation [34]. Astrocytic end-feet project toward the vasculature, forming polarized connections that support barrier function through proteins including aquaporin IV and the dystroglycan-dystrophin complex [34].
Transport Pathways
Substances cross the BBB via specific biological routes with distinct structural requirements:
- Transmembrane Diffusion: The primary pathway for most CNS drugs, this passive transport mechanism depends critically on a compound's physicochemical properties, favoring molecules with low molecular weight (<400-600 Da) and moderate lipid solubility [107] [109] [108].
- Carrier-Mediated Transport (CMT): Specialized influx transporters (e.g., GLUT1 for glucose, LAT1 for large neutral amino acids) shuttle essential nutrients across the BBB via saturable carrier systems that operate roughly 10-fold faster than transmembrane diffusion [107] [108].
- Receptor-Mediated Transcytosis (RMT): Vesicular transport mechanisms for macromolecules including insulin (via insulin receptors), transferrin (via transferrin receptors), and lipoproteins (via LDL receptors) enable selective large molecule passage [107] [34].
- Adsorptive-Mediated Transcytosis (AMT): Charge-based interactions between cationic molecules and anionic membrane microdomains facilitate non-specific uptake of certain peptides and proteins [34] [108].
- Efflux Transport: ATP-binding cassette transporters including P-glycoprotein (P-gp), breast cancer resistant protein (BCRP), and multidrug resistance-associated proteins (MRPs) actively extrude substrates back into the circulation, significantly limiting brain penetration for many compounds [107] [110].
The following diagram illustrates the primary cellular components and transport pathways at the Blood-Brain Barrier:
Lipophilicity and Its Critical Role in BBB Penetration
Fundamental Principles
Lipophilicity, quantitatively expressed as the partition coefficient (log P) between octanol and water, represents one of the most critical physicochemical parameters influencing passive diffusion across the BBB [5] [6] [15]. This property determines a molecule's ability to traverse lipid membranes while maintaining sufficient aqueous solubility for systemic delivery [6]. The relationship between lipophilicity and BBB permeability follows a well-established parabolic pattern, with optimal brain penetration typically occurring at log P values between 1.5 and 3.5 [5] [109]. Compounds with excessive lipophilicity (log P > 5) often demonstrate poor aqueous solubility, increased metabolic clearance, and heightened plasma protein binding, thereby reducing bioavailability and brain exposure [5] [6].
Lipophilicity-ADMET Relationships
The influence of lipophilicity extends beyond permeability to encompass comprehensive ADMET properties:
- Absorption: Moderate lipophilicity enhances passive transcellular absorption through biological membranes, including the gastrointestinal epithelium and BBB [15] [49].
- Distribution: Increased lipophilicity elevates volume of distribution and tissue binding, potentially prolonging half-life but also increasing accumulation in peripheral tissues and fat deposits [5] [15].
- Metabolism: Highly lipophilic compounds demonstrate faster metabolic turnover through cytochrome P450 systems, potentially reducing systemic exposure and necessitating higher dosing [5] [6].
- Excretion: Lipophilic drugs may undergo biliary excretion and enterohepatic recirculation, complicating elimination kinetics [15].
- Toxicity: Excessive lipophilicity correlates with promiscuous target binding, mitochondrial toxicity, and phospholipidosis, increasing safety liabilities [5] [6].
The following table summarizes key property relationships for optimal CNS penetration:
Table 1: Optimal Physicochemical Properties for BBB Penetration
| Property | Optimal Range | Impact on BBB Penetration | ADMET Consequences |
|---|---|---|---|
| Lipophilicity (log P) | 1.5-3.5 [5] [109] | Determines passive diffusion rate | Values >5: Poor solubility, rapid metabolism, tissue accumulation [5] [6] |
| Molecular Weight | <400-500 Da [107] [109] | Inverse relationship with permeability | Higher MW reduces diffusion coefficient and membrane transit |
| Hydrogen Bond Donors | ≤3 [109] | Reduced H-bonding capacity enhances permeability | Excessive HBD decreases membrane penetration |
| Hydrogen Bond Acceptors | ≤7 [109] | Limited polar surface area improves diffusion | Increased PSA correlates with reduced BBB penetration |
| P-gp Substrate | No [110] | Avoids active efflux transport | P-gp substrates demonstrate reduced brain-to-plasma ratios |
Experimental Methodologies for Assessing BBB Penetration
Lipophilicity Determination Methods
Accurate measurement of lipophilicity provides critical data for structure-permeability relationships:
Chromatographic Techniques (RP-TLC/RP-HPLC): Reverse-phase thin-layer chromatography and high-performance liquid chromatography determine the chromatographic parameter RM⁰, which correlates with octanol-water partition coefficients. These methods offer high throughput, minimal compound requirements, and excellent reproducibility compared to traditional shake-flask approaches [5] [15] [49]. Mobile phases typically comprise acetone-TRIS buffer (pH 7.4) mixtures with varying organic modifier concentrations [15] [49].
Shake-Flask Method: The classical approach directly measures partition coefficients between n-octanol and aqueous buffers, providing definitive log P values within a range of -2 to 4. Despite time-consuming procedures and requirements for compound purity, this method remains the gold standard for validation [15].
Calculated log P Values: Computational programs including iLOGP, XLOGP3, WLOGP, MLOGP, and SILICOS-IT apply diverse algorithms to predict lipophilicity from molecular structure [5] [15] [49]. These tools enable rapid screening during early discovery phases, though accuracy varies between chemical classes.
In Silico ADMET Prediction Platforms
Computational tools have become indispensable for predicting BBB penetration and pharmacokinetic properties:
- SwissADME: Provides comprehensive pharmacokinetic profiling including BBB permeability predictions, gastrointestinal absorption, and drug-likeness assessments [5] [15] [49].
- pkCSM: Predicts absorption, distribution, metabolism, excretion, and toxicity parameters from molecular structure [15] [49].
- SwissTargetPrediction: Estimates potential protein targets and off-target interactions [5].
- ADMETlab: Offers integrated ADMET property evaluation with consensus scoring [15].
The following workflow diagram illustrates a typical experimental protocol for evaluating BBB penetration:
In Vitro and In Vivo Evaluation Methods
- In Situ Brain Perfusion: This technique directly measures BBB permeability by perfusing test compounds through the carotid artery followed by brain capillary depletion, providing precise quantification of uptake clearance [110].
- Brain-to-Plasma Ratio (Kp): The most widely used in vivo parameter compares total drug concentrations in brain and plasma at steady state. While easily measurable, Kp can be misleading without considering free drug fractions [110].
- Microdialysis: This method measures unbound drug concentrations in brain interstitial fluid, providing the most pharmacologically relevant exposure data but requiring specialized technical expertise [110].
- P-gp Efflux Assays: Cell systems overexpressing P-glycoprotein (e.g., MDCK-MDR1, Caco-2) identify substrates for this critical efflux transporter, enabling early elimination of compounds with poor brain penetration potential [110].
Strategic Approaches to Enhance BBB Penetration
Passive Diffusion Optimization
Structural modification to fine-tune physicochemical properties represents the most direct approach for improving passive diffusion:
- Prodrug Strategies: Chemical derivatization to increase lipophilicity facilitates BBB penetration, with subsequent enzymatic conversion to active drug within the CNS. The classic example of heroin (diacetylmorphine) demonstrates this principle—acylation significantly enhances lipid solubility and brain uptake compared to morphine, with subsequent deacetylation to active metabolites [109].
- Molecular Weight Optimization: Reducing molecular mass below 500 Da enhances diffusion coefficients and membrane transit [107] [109].
- Hydrogen Bonding Minimization: Decreasing the number of hydrogen bond donors and acceptors reduces polar surface area, improving membrane permeability [109].
- pKa Modulation: Adjusting ionization potential to maximize uncharged species at physiological pH enhances lipid membrane partitioning [6].
Targeted Delivery Technologies
Advanced delivery systems exploit endogenous transport mechanisms to overcome BBB limitations:
- Nanoparticle Systems: Polymeric, lipid-based, and inorganic nanoparticles protect therapeutic cargo and can be surface-functionalized with targeting ligands to enhance brain delivery [107] [34]. These systems provide sustained release profiles and potential for bypassing efflux transporters [107].
- Monoclonal Antibody Approaches: Bispecific antibodies with one binding site targeting transferrin receptor (TfR) or insulin receptor (IR) and another engaging therapeutic targets facilitate receptor-mediated transcytosis [107]. Importantly, low-affinity TfR binding maximizes brain uptake while minimizing peripheral sequestration [107].
- Peptide Vectors: Cell-penetrating peptides and receptor-targeting peptides (e.g., Angiopep-2 targeting LRP1) enable shuttle-mediated transport of conjugated therapeutics [107]. The ANG-1005 conjugate (paclitaxel-Angiopep-2) demonstrates significantly improved BBB penetration compared to unconjugated paclitaxel [107].
Transient Barrier Modulation
- Chemical Disruption: Bradykinin analogs (e.g., RMP-7) and alkylglycerols temporarily increase paracellular permeability through tight junction modulation [108].
- Focused Ultrasound: Microbubble-assisted ultrasound techniques locally and reversibly disrupt the BBB through acoustic cavitation effects, enabling targeted drug delivery [108].
- Osmotic Disruption: Intra-arterial hyperosmotic solutions (e.g., mannitol) transiently shrink endothelial cells and open tight junctions, though this approach affects the entire brain and carries significant safety liabilities [108].
The Scientist's Toolkit: Essential Research Reagents and Methods
Table 2: Key Research Reagent Solutions for BBB Penetration Studies
| Reagent/Assay | Function | Application Context |
|---|---|---|
| MDCK-MDR1 Cells | In vitro permeability model with human P-gp overexpression | Efflux transporter substrate identification [110] |
| Brain Homogenate Binding | Determination of free fraction in brain tissue (fu,brain) | Correction of total brain concentrations to unbound levels [110] |
| In Situ Brain Perfusion | Direct measurement of BBB permeability | Quantification of uptake clearance without confounding systemic factors [110] |
| RP-TLC Chromatography | Experimental lipophilicity determination (RM⁰) | High-throughput assessment of partition coefficients [5] [15] |
| SwissADME Platform | In silico prediction of ADMET properties | Virtual screening of BBB permeability and drug-likeness [5] [15] |
| P-gp Inhibitors (e.g., Elacridar) | Chemical inhibition of efflux transport | Mechanistic studies on transporter effects [110] |
Optimizing CNS drug penetration requires meticulous balancing of lipophilicity within the context of comprehensive ADMET properties. Successful candidates typically demonstrate moderate lipophilicity (log P 1.5-3.5), molecular weight <500 Da, limited hydrogen bonding capacity, and minimal recognition by efflux transporters, particularly P-glycoprotein. Beyond these fundamental properties, emerging strategies including nanoparticle delivery systems, bispecific antibodies, and transient barrier modulation offer promising approaches for challenging therapeutics. A multidisciplinary methodology integrating computational prediction, robust in vitro screening, and appropriate in vivo validation provides the most effective path toward developing CNS therapeutics with optimal exposure and efficacy. The continuing evolution of BBB penetration strategies will undoubtedly expand the therapeutic landscape for neurological disorders, addressing one of the most persistent challenges in pharmaceutical development.
Strategic Molecular Modifications to Modulate Lipophilicity
Lipophilicity, a compound's ability to dissolve in non-polar solvents, represents one of the most fundamental physicochemical parameters in drug discovery and development. It is typically quantified as the logarithm of the n-octanol/water partition coefficient (logP) or distribution coefficient (logD), which measure the equilibrium concentration of a compound between organic and aqueous phases [5] [1]. This property exerts profound influence on a molecule's behavior in biological systems, directly affecting its absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile [1] [15] [111]. As noted in recent studies of anticancer diazaphenothiazines, "lipophilicity is one of the key properties of a potential drug that determines the solubility, the ability to penetrate through cell barriers, and transport to the molecular target" [5].
The relationship between lipophilicity and bioavailability follows a non-linear pattern, with an optimal range (typically logP 1-3) being essential for therapeutic efficacy [111]. Excessively low lipophilicity often results in poor membrane permeability, while highly lipophilic compounds tend to exhibit inadequate aqueous solubility, increased metabolic clearance, and potential toxicity issues [5] [1] [111]. As pharmaceutical pipelines increasingly incorporate highly lipophilic compounds, strategic molecular modification to fine-tune this parameter has become an indispensable component of rational drug design [50] [112].
Lipophilicity-ADMET Relationships: A Molecular Basis for Design
The following diagram illustrates the complex relationships between lipophilicity and key ADMET properties that drug developers must balance during molecular optimization:
Figure 1: The Dual-Faced Impact of Lipophilicity on ADMET Properties. Lipophilicity exhibits complex, often opposing relationships with key pharmacokinetic parameters, necessitating careful optimization within a defined optimal range (typically logP 1-3) to balance favorable membrane permeability with acceptable solubility, metabolic stability, and toxicity profiles [5] [1] [111].
Key ADMET Considerations in Lipophilicity Optimization
The impact of lipophilicity on biological disposition manifests through several critical mechanisms:
Absorption and Permeability: Lipophilicity fundamentally governs a compound's ability to passively diffuse across biological membranes, including the gastrointestinal epithelium and the blood-brain barrier [113] [1]. As noted in studies of tacrine-based cholinesterase inhibitors, "compounds with higher lipophilicity tend to bind more strongly to plasma proteins which can reduce the free fraction of the drug available to reach the target tissues" [114].
Metabolic Stability: Increasing lipophilicity typically correlates with enhanced metabolic vulnerability, particularly to cytochrome P450-mediated oxidation, potentially reducing systemic exposure [1] [111]. Recent analyses of protein-protein interaction inhibitors revealed that compounds with higher lipophilicity values (logP > 4) frequently exhibit unfavorable metabolic profiles and increased promiscuity [50].
Solubility-Bioavailability Tradeoff: Perhaps the most significant challenge lies in balancing the opposing effects of lipophilicity on solubility and permeability. Research on quinoline-1,4-quinone hybrids demonstrates that "too high values of lipophilicity contribute to the immobilization of the compound" within lipid bilayers, paradoxically reducing absorption despite favorable partitioning [49].
Strategic Molecular Modification Approaches
The following workflow outlines a systematic approach to lipophilicity optimization, integrating computational prediction, strategic molecular design, and experimental validation:
Figure 2: Integrated Workflow for Lipophilicity Optimization. This systematic approach combines computational prediction with strategic molecular modifications and experimental validation to achieve targeted lipophilicity adjustments while maintaining optimal ADMET properties [15] [111] [115].
Core Molecular Modification Strategies
Table 1: Strategic Molecular Modifications for Lipophilicity Optimization
| Modification Strategy | Structural Change | Impact on logP | Key Considerations |
|---|---|---|---|
| Bioisosteric Replacement | Exchange of atoms/groups with similar physicochemical properties | Variable (typically -1 to +1 log unit) | Maintains target affinity while modulating properties; halogen replacement with trifluoromethyl groups reduces logP by ~0.5-1 unit [113] [115] |
| Alkyl Chain Optimization | Lengthening/shortening or branching of aliphatic chains | +/- 0.5-0.7 per methylene group | Critical for balancing permeability and solubility; 1,9-diazaphenothiazines showed 1.83-3.13 logP range with different alkyl substituents [5] [111] |
| Ring System Modification | Introduction/removal of aromatic/aliphatic rings or heteroatoms | Variable (typically +1.5 to +3 per aromatic ring) | Incorporating nitrogen heteroatoms in quinoline-1,4-quinone hybrids significantly reduced lipophilicity [49] [115] |
| Polar Group Introduction | Addition of hydroxyl, amine, carbonyl, or sulfonyl groups | -0.5 to -2.5 per group | Improves aqueous solubility but may reduce membrane permeability; strategic placement avoids disruption of pharmacophore [111] [115] |
| Molecular Hybridization | Covalent linking of pharmacophores from distinct bioactive molecules | Additive or synergistic effects | Creates multi-targeting compounds; requires careful optimization of linker length and flexibility [115] |
Strategic molecular modification requires careful consideration of structure-liophilicity relationships to achieve precise adjustments while maintaining pharmacological activity:
Bioisosteric Replacement: This approach involves substituting molecular fragments with isosteres that possess similar physicochemical properties but different lipophilicity. Studies on tacrine derivatives demonstrated that strategic fluorine substitution effectively modulated logP while preserving cholinesterase inhibitory activity [114]. The replacement of lipophilic groups with polar bioisosteres (e.g., tetrazole for carboxylic acid) can significantly reduce logP while maintaining key molecular interactions [115].
Alkyl Chain Optimization: Systematic variation of alkyl substituents represents one of the most straightforward approaches to fine-tune lipophilicity. In 1,9-diazaphenothiazines, the length and branching of alkyl chains at position 10 produced logP values ranging from 1.83 (methyl) to 3.13 (benzyl), enabling researchers to identify analogs with optimal membrane permeability and solubility profiles [5]. Recent work on diquinothiazines further confirmed that dialkylaminoalkyl substituents can be strategically optimized to achieve target lipophilicity ranges [15].
Polar Group Incorporation: Introducing hydrogen bond donors or acceptors represents an effective strategy for reducing excessive lipophilicity. Research on quinoline-1,4-quinone hybrids demonstrated that "introduction of the nitrogen atom reduced the lipophilicity depending on the position at the 5,8-quinolinedione moiety" [49]. However, this approach requires careful optimization, as excessive polarity can compromise membrane permeability and central nervous system exposure [113].
Experimental Determination of Lipophilicity
Accurate measurement of lipophilicity parameters is essential for validating computational predictions and guiding structural optimization. The following table compares principal experimental methods for lipophilicity determination:
Table 2: Comparison of Experimental Methods for Lipophilicity Determination
| Method | Measurement Range (logP) | Sample Requirements | Throughput | Key Advantages | Limitations |
|---|---|---|---|---|---|
| Shake-Flask | -2 to 4 | High purity, ~1-10 mg | Low (hours to days) | Gold standard, direct measurement, applicable to ionizable compounds | Time-consuming, requires high compound purity, limited range [112] |
| RP-TLC | 0 to 6 | Moderate purity, μg scale | Medium-High | Cost-effective, minimal sample requirement, high throughput | Indirect measurement, requires reference compounds [5] [49] [15] |
| RP-HPLC | 0 to >6 | Low-moderate purity, μg scale | High | Broad measurement range, robust, automated | Indirect measurement, method development required [114] [112] |
| Computational Prediction | Unlimited | Structure only | Very High | Instant prediction, no compound needed, guides design | Accuracy varies by algorithm, limited for novel scaffolds [5] [50] |
Detailed Experimental Protocols
RP-TLC Method for Lipophilicity Screening
Reversed-phase thin-layer chromatography has emerged as a preferred method for rapid lipophilicity screening during early drug discovery due to its simplicity, cost-effectiveness, and minimal sample requirements [5] [49] [15]. The standard protocol involves:
- Stationary Phase: RP-18W F254s TLC plates (e.g., Merck, Darmstadt, Germany) [15] [114].
- Mobile Phase: Binary mixtures of acetone or methanol with TRIS buffer (0.2 M, pH 7.4) or water acidified with formic acid [49] [114]. Organic modifier concentrations typically range from 50% to 80% in 5% increments.
- Sample Application: 1.0-5.0 μL of ethanolic solutions (0.5-1.0 mg/mL) applied as spots using micropipettes [49] [114].
- Detection: UV light (254 nm) or iodine vapor visualization [49].
- Data Analysis: Retardation factor (Rf) values are converted to RM values using the equation RM = log(1/Rf - 1). The chromatographic lipophilicity parameter (RM0) is determined by extrapolating RM values to zero organic modifier concentration using the equation RM = RM0 + bC, where C is the concentration of organic modifier [49] [15].
This method was successfully applied to determine the lipophilicity of 1,9-diazaphenothiazines, demonstrating excellent correlation coefficients (0.968-0.999) between acetone concentration and retardation factors [5] [49].
RP-HPLC Method for Accurate logP Determination
Reversed-phase high-performance liquid chromatography provides more accurate logP determination, particularly for compounds with high lipophilicity (logP > 5) [112]. Two established approaches include:
Rapid Screening Method (Method 1):
- Reference Compounds: A calibration set of 6 compounds with known logP values (e.g., 4-acetylpyridine, acetophenone, chlorobenzene, ethylbenzene, phenanthrene, triphenylamine) covering logP range 0.5-5.7 [112].
- Chromatographic Conditions: C18 column (e.g., 4.6 × 150 mm, 5 μm), methanol-water or acetonitrile-water gradient, flow rate 1.0 mL/min, detection at 254 nm [112].
- Data Analysis: Capacity factors (k) are calculated from retention times. A calibration curve is constructed by plotting logk against known logP values of reference compounds. The logP of test compounds is calculated from their capacity factors using the established linear relationship: logP = a × logk + b [112].
High-Accuracy Method (Method 2):
- This method replaces logk with logkw (capacity factor in pure aqueous phase) to eliminate interference from organic modifiers [112].
- Data Analysis: Retention times are measured at three different organic modifier concentrations. For each compound, the relationship between logk and organic modifier content (φ) is established: logk = Sφ + logkw. The logkw values are used to construct a more accurate calibration curve: logP = a × logkw + b [112].
Method 1 provides rapid screening (≤30 minutes per compound) with good accuracy (R² = 0.97), while Method 2 offers superior predictive ability (R² = 0.996) with longer analysis time (2-2.5 hours) [112].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Lipophilicity Studies
| Category | Specific Items | Application Purpose | Technical Notes |
|---|---|---|---|
| Chromatography Materials | RP-18W F254s TLC plates | Stationary phase for RP-TLC | Aluminum-backed, 10 × 10 cm plates; enable high-throughput screening [49] [114] |
| C18 HPLC columns (4.6 × 150 mm, 5 μm) | Stationary phase for RP-HPLC | Standard dimensions for logP determination; provides reproducible retention [112] | |
| Mobile Phase Components | HPLC-grade methanol, acetonitrile | Organic modifiers for mobile phase | Methanol preferred for hydrogen bonding similarity to n-octanol [112] |
| TRIS buffer (0.2 M, pH 7.4) | Aqueous component simulating physiological pH | Maintains consistent pH environment during measurements [49] [15] | |
| Reference Compounds | 4-Acetylpyridine (logP 0.5), acetophenone (logP 1.7) | Low-mid lipophilicity calibrants | Essential for establishing calibration curves [112] |
| Chlorobenzene (logP 2.8), ethylbenzene (logP 3.2) | Mid-range lipophilicity calibrants | Cover critical range for drug-like molecules [112] | |
| Phenanthrene (logP 4.5), triphenylamine (logP 5.7) | High lipophilicity calibrants | Enable measurement extension to logP >5 [112] | |
| Software Tools | SwissADME, pkCSM | In silico logP prediction | Web-based platforms providing multiple algorithm consensus [5] [15] [50] |
| AutoDock Vina, Gaussian | Molecular modeling and docking | Assess binding interactions post-modification [49] [115] |
Strategic molecular modification to modulate lipophilicity represents a critical capability in modern drug discovery, directly impacting a compound's ADMET profile and ultimate therapeutic utility. The integration of computational prediction with experimental validation through chromatographic methods provides a robust framework for achieving target lipophilicity ranges. As demonstrated in studies across diverse chemical classes—from 1,9-diazaphenothiazines to quinolone-1,4-quinone hybrids—systematic application of bioisosteric replacement, alkyl chain optimization, and polar group incorporation enables precise fine-tuning of this fundamental property. The continued refinement of these strategies, coupled with advances in high-throughput screening and computational modeling, will further enhance our ability to optimize drug candidates for improved bioavailability and therapeutic efficacy.
Employing Prodrug Strategies and Formulation to Overcome ADMET Hurdles
Lipophilicity, quantitatively represented as the partition coefficient (log P), is a fundamental physicochemical property that critically influences a drug's Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET). It determines a molecule's ability to dissolve in aqueous versus lipid environments, thereby governing its behavior in biological systems. Drugs typically require a balance; excessive lipophilicity (log P > 5) is associated with poor aqueous solubility, high metabolic turnover, and tissue accumulation, while insufficient lipophilicity can limit membrane permeability [5] [14]. According to Lipinski's "Rule of Five," an estimated log P value below 5 is a key criterion for oral drug-likeness, with most successful drugs exhibiting log P values between 0 and 3 for optimal oral bioavailability [5] [14]. For specific targets like the central nervous system (CNS), an ideal log P of approximately 2 is often necessary for effective blood-brain barrier (BBB) penetration [14] [116]. Consequently, modulating lipophilicity is a primary objective of prodrug strategies, which use bioreversible derivatives of active drugs to temporarily alter physicochemical properties and overcome ADMET barriers [117] [118].
The Prodrug Approach: Rationale and Strategic Implementation
Prodrugs are pharmacologically inactive molecules designed to undergo enzymatic or chemical transformation in vivo to release the active parent drug. This approach has evolved from a last-resort intervention to an integral component of modern drug design, accounting for approximately 12% of all FDA-approved new small-molecular entities and 10% of marketed drugs worldwide [117] [118]. The strategic application of prodrug technology can resolve a wide array of development challenges:
- Improving Water Solubility: Enhancing dissolution and bioavailability for highly lipophilic compounds.
- Enhancing Membrane Permeability: Increasing absorption and tissue penetration for polar drugs.
- Achieving Site-Specific Targeting: Exploiting unique enzymes or transporters at the target site for localized activation.
- Mitigating Pre-systemic Metabolism: Protecting the drug from enzymatic degradation before it reaches systemic circulation.
- Reducing Toxicity and Side Effects: Minimizing exposure to the active drug in non-target tissues.
The following conceptual framework outlines the core objectives and design logic of a prodrug development program:
Lipophilicity and ADMET: Quantitative Relationships and Experimental Assessment
The influence of lipophilicity on key ADMET parameters is well-established. Understanding these relationships allows scientists to predict compound behavior and design better prodrugs. The following table summarizes the core impacts of log P on drug disposition.
Table 1: Influence of Lipophilicity on Key ADMET Properties
| ADMET Parameter | Impact of Low Lipophilicity (log P < 0) | Impact of High Lipophilicity (log P > 5) | Optimal Range (General) |
|---|---|---|---|
| Absorption (Oral) | Good solubility but potentially poor permeability due to inability to cross lipid membranes [14]. | Good permeability but poor solubility and dissolution, limiting absorption [5] [14]. | log P 0–3 for balanced solubility and permeability [14]. |
| Distribution (Volume) | Limited tissue penetration, low volume of distribution [14]. | High tissue accumulation and plasma protein binding, leading to large volume of distribution and potential toxicity [5] [14]. | Dependent on target tissue; ~2 for BBB penetration [5] [14]. |
| Metabolism | Often lower metabolic clearance. | High metabolic turnover by cytochrome P450 enzymes [5]. | Moderate log P to avoid being a substrate for metabolizing enzymes. |
| Excretion | Typically rapid renal excretion. | Biliary excretion and potential for prolonged half-life due to tissue storage [5]. | Moderate log P to avoid excessive accumulation. |
| Toxicity | Generally lower, but off-target effects possible. | Increased risk of nonspecific toxicity and organ accumulation [5] [15]. | Moderate log P to minimize nonspecific binding. |
Experimental Protocols for Determining Lipophilicity
Accurate determination of lipophilicity is crucial for rational prodrug design. The following experimental methods are standard in the field:
Shake-Flask Method (Reference Standard): This is the classic benchmark technique for determining log P.
- Methodology: The compound is partitioned between n-octanol (lipophilic phase) and an aqueous buffer (hydrophilic phase). The mixture is shaken vigorously to reach equilibrium and then allowed to separate. The concentration of the compound in each phase is quantified using a suitable analytical technique (e.g., UV spectrophotometry, HPLC).
- Calculation: log P = log10(Concentrationinoctanol / Concentrationinwater).
- Limitations: It is labor-intensive, requires highly pure compounds, and is less accurate for compounds with extreme log P values [14] [15].
Reversed-Phase Chromatography (RP-TLC/RP-HPLC): Chromatographic methods are widely used due to their speed, reproducibility, and minimal compound requirement.
- RP-TLC Protocol:
- Stationary Phase: C18-modified silica plates.
- Mobile Phase: A binary mixture of a water-miscible organic modifier (e.g., acetone, methanol) and an aqueous buffer (e.g., TRIS buffer pH 7.4) [5] [15].
- Measurement: The retention factor (Rₘ) is calculated from the compound's migration distance. Rₘ values are determined for several mobile phase compositions and extrapolated to 0% organic modifier to obtain Rₘ⁰, which correlates directly with lipophilicity [5] [15].
- RP-HPLC Protocol:
- Stationary Phase: C18 column.
- Mobile Phase: Gradient or isocratic elution with water-organic modifier mixtures.
- Measurement: The retention time (or capacity factor, k) is measured. The log k value extrapolated to 0% organic modifier (log k₀) serves as a chromatographic lipophilicity index [14].
- RP-TLC Protocol:
The typical workflow for the chromatographic determination of lipophilicity is as follows:
Prodrug Design Strategies to Modulate Lipophilicity and Overcome ADMET Hurdles
Prodrug design can be categorized into two main approaches: traditional strategies aimed at global optimization of physicochemical properties, and modern strategies that incorporate cellular and molecular targeting.
Traditional Prodrug Strategies: Esterification
Ester prodrugs are the most prevalent strategy for temporarily increasing the lipophilicity of polar drugs containing hydroxyl, phenol, or carboxylate groups.
- Mechanism: These polar groups are masked with lipophilic promoieties (e.g., alkyl chains, pivaloyl) via an ester bond. This modification significantly reduces hydrogen bonding capacity and increases log P, thereby enhancing passive diffusion across biological membranes like the intestinal mucosa and the BBB [116] [117].
- Activation: The ester bond is cleaved in vivo by ubiquitous esterases (e.g., carboxylesterases, acetylcholinesterase) to release the active parent drug.
- Case Study - Brain Delivery: The polar anticancer drug methotrexate (MTX) has poor BBB penetration. The synthesis of a dihexyl ester prodrug decreased unspecific hydrolysis and increased the brain-to-plasma ratio by 6-fold, significantly enhancing efficacy and reducing off-target effects [116].
Modern Prodrug Strategies: Carrier-Mediated Transport
This advanced approach designs prodrugs to be substrates for specific nutrient transporters expressed on biological barriers, such as the BBB.
- Mechanism: The drug is conjugated to a promoiety that mimics an endogenous substrate for transporters like GLUT1 (glucose), LAT1 (large neutral amino acids), or PEPTI (peptides). This enables active, carrier-mediated uptake of the prodrug into the target tissue [116] [117].
- Activation: Following transport, the prodrug is cleaved by intracellular enzymes to release the active drug.
- Advantage: This strategy can achieve higher and more selective tissue accumulation compared to passive diffusion alone.
The journey of a transporter-targeted prodrug from administration to activation illustrates this sophisticated strategy:
Advanced Formulation: Nano-formulated Prodrugs
Combining prodrug chemistry with nanotechnology can further enhance drug delivery, particularly for challenging agents like the potent camptothecin analogue SN38.
- Strategy: A dual-modified SN38 prodrug was created by esterifying both its 10-OH and 20-OH positions with polyunsaturated fatty acids (PUFAs) like linoleic acid. This drastically increased lipophilicity and compatibility with biodegradable amphiphilic block polymers (e.g., PEG-b-PCL) [119].
- Formulation: The prodrugs were co-assembled with the polymers into stable nanoparticles for intravenous injection.
- Outcome: This nano-formulated prodrug demonstrated superior stability, sustained drug release, enhanced pharmacokinetics, and markedly higher antitumor efficacy in pancreatic cancer models compared to the clinical prodrug irinotecan [119].
In Silico and Experimental Tools for Prodrug Evaluation
The prodrug development pipeline relies on a combination of computational predictions and experimental validation.
In Silico Prediction Tools
Computational tools are indispensable for early-stage screening and property prediction. Table 2: Key In Silico Platforms for ADMET and Prodrug Profiling
| Tool Name | Primary Function | Utility in Prodrug Design |
|---|---|---|
| SwissADME [5] [15] | Predicts drug-likeness, physicochemical properties, and ADME parameters. | Rapidly calculates log P (using iLOGP, XLOGP3, etc.), solubility, and assesses compliance to rules like Lipinski's. |
| pkCSM [15] [20] | Predicts a wide range of pharmacokinetic and toxicity parameters. | Useful for forecasting ADMET profiles of both parent drug and prodrug candidate. |
| SwissTargetPrediction [5] | Predicts the most probable protein targets of a small molecule. | Helps understand potential off-target interactions of the prodrug and its promoiety. |
| PharmaBench [20] | A large-scale, curated benchmark dataset for building ADMET prediction AI models. | Provides high-quality data for training and validating custom predictive models in drug discovery projects. |
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key materials and reagents used in prodrug research and lipophilicity assessment.
Table 3: Essential Research Reagent Solutions for Prodrug and ADMET Studies
| Reagent / Material | Function and Application |
|---|---|
| C18-modified Silica Plates/Columns | The stationary phase for Reversed-Phase (RP) TLC and HPLC, simulating the lipophilic environment for determining lipophilicity indices (Rₘ⁰, log k₀) [5] [14]. |
| n-Octanol & Aqueous Buffers | The two-phase system used in the classic shake-flask method for direct experimental log P determination [14]. |
| Carboxylesterase Enzymes | Key hydrolytic enzymes used in in vitro studies to simulate the metabolic activation of ester-based prodrugs [116] [119]. |
| Polymeric Nanocarriers (e.g., PEG-b-PCL, PEG-b-PLA) | Biodegradable amphiphilic block copolymers used to formulate lipophilic prodrugs into stable, injectable nanoparticles, improving pharmacokinetics and targeting [119]. |
| Linoleic Acid / Docosahexaenoic Acid (DHA) | Polyunsaturated fatty acids (PUFAs) used as lipophilic promoieties in prodrug synthesis to enhance compatibility with drug delivery systems and modulate release profiles [119]. |
The strategic integration of prodrug design and advanced formulation technologies presents a powerful approach to overcoming pervasive ADMET hurdles. By systematically modulating lipophilicity through chemical modification—such as esterification for enhanced permeability or transporter-targeted designs for site-specific delivery—scientists can significantly improve the therapeutic index of drug candidates. The continued synergy between experimental chromatographic techniques, sophisticated in silico prediction tools, and innovative nanocarrier systems will undoubtedly accelerate the development of safer and more effective prodrug-based medicines, solidifying this approach as a cornerstone of modern medicinal chemistry and drug delivery.
Multi-parameter optimization (MPO) represents a fundamental paradigm shift in modern drug discovery, addressing the critical challenge of balancing often-conflicting compound properties to identify high-quality drug candidates. This technical guide examines MPO methodologies with particular emphasis on lipophilicity, a principal physicochemical parameter with profound influence on absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles. By integrating computational predictions with experimental validation strategies, researchers can systematically navigate chemical space to identify compounds with optimal lipophilicity balanced against potency, solubility, and safety parameters. This whitepaper provides detailed protocols, computational frameworks, and strategic approaches for implementing effective MPO in pharmaceutical development, enabling researchers to reduce late-stage attrition and advance higher-quality drug candidates.
Lipophilicity, quantitatively expressed as the partition coefficient (log P) or distribution coefficient (log D), is one of the most critical physicochemical parameters in drug design due to its profound influence on pharmacokinetic behavior [11]. As a key determinant of molecular behavior in biological systems, lipophilicity affects a drug candidate's ability to cross cell membranes, reach therapeutic targets, and avoid accumulation in fatty tissues [11]. The fundamental relationship between lipophilicity and ADMET properties creates both opportunities and challenges for drug designers seeking to optimize multiple parameters simultaneously.
The MPO Imperative in Modern Drug Discovery Contemporary small molecule drug discovery necessitates rapid and simultaneous optimization of numerous parameters, extending far beyond traditional potency measures [120]. A successful, efficacious, and safe drug must achieve a delicate balance of properties, including target potency, appropriate ADMET characteristics, and an acceptable safety profile [121]. This balancing of often conflicting requirements represents a major challenge that MPO approaches aim to address through systematic frameworks that consider multiple parameters concurrently rather than sequentially [121] [120].
Lipophilicity as a Master Parameter Lipophilicity serves as a central parameter in MPO frameworks due to its extensive correlations with diverse ADMET endpoints [11]. Compounds with moderate lipophilicity typically demonstrate better absorption through cell membranes, while excessive lipophilicity can lead to poor solubility, increased metabolic clearance, and higher toxicity risk [11] [122]. Understanding and controlling lipophilicity within optimal ranges therefore provides a powerful strategy for improving compound quality and developmental viability.
Lipophilicity and ADMET Relationships: Quantitative Foundations
The following table summarizes key relationships between lipophilicity and critical ADMET parameters, providing quantitative guidance for MPO target setting:
Table 1: Lipophilicity Relationships with ADMET Properties and Optimization Targets
| ADMET Parameter | Relationship with Lipophilicity | Optimal Range | Key Considerations |
|---|---|---|---|
| Absorption | Bell-shaped relationship; moderate log P enhances passive diffusion | log P 1-3 [122] | Excessive lipophilicity (>5) reduces solubility and absorption [122] |
| Distribution | Increased tissue penetration and volume of distribution with higher log P | Compound-dependent | High lipophilicity may cause unwanted CNS penetration or adipose tissue accumulation |
| Metabolism | Higher susceptibility to cytochrome P450 metabolism with increased log P | Minimize metabolic hotspots | Lipophilic compounds often show faster metabolic clearance |
| Toxicity | Correlated with hERG inhibition and general toxicity risk | log P < 5 [123] | Promiscuity and toxicity risks increase significantly beyond log P > 5 |
| Solubility | Inverse relationship; higher log P reduces aqueous solubility | log P 1-3 for acceptable solubility [122] | Critical for oral bioavailability; must balance with permeability |
Experimental Lipophilicity Determination Methods Accurate lipophilicity assessment employs both traditional and chromatographic techniques, each with distinct advantages and limitations:
Shake-Flask Method: The classic reference technique recommended by OECD, which involves direct measurement of partition coefficients between octanol and water phases. This method provides accurate log P values in the range of -2 to 4 but requires relatively large amounts of pure compounds and is time-consuming [11].
Chromatographic Methods: Reversed-phase chromatographic techniques (RP-TLC and RP-HPLC) provide indirect lipophilicity measurements with several practical advantages. These methods require smaller sample amounts and offer faster analysis times while maintaining reasonable accuracy (±1 unit compared to shake-flask values) [11]. The chromatographic parameter R₀ is widely used as an experimental lipophilicity index.
Computational Lipophilicity Prediction Multiple in silico approaches have been developed for rapid log P estimation, especially valuable during early discovery stages:
Table 2: Computational Methods for Lipophilicity Prediction
| Method Type | Representative Tools | Key Features | Accuracy Considerations |
|---|---|---|---|
| Fragment-Based | iLOGP, XLOGP3 | Sum of fragment contributions with correction factors | iLOGP showed strong correlation with chromatographic values in diquinothiazine studies [11] |
| Property-Based | MLOGP | Uses molecular properties like polarity and surface area | Fast calculation but may lack chemical context |
| Machine Learning | SILCOS-IT, milogP | Pattern recognition from training sets | Performance depends on training data quality and chemical space similarity |
MPO Methodologies: Frameworks for Balanced Compound Design
Multiple computational frameworks have been developed to address the challenges of MPO in drug discovery, integrating lipophilicity with other critical parameters:
MPO Methodologies Overview
Probabilistic Scoring Approaches Probabilistic scoring methods assess the likelihood of compound success by simultaneously considering multiple properties according to their desired values and relative importance [122]. These approaches uniquely incorporate uncertainty associated with each property prediction, generating a composite score between 0 and 1 that estimates the compound's probability of success [122]. This method is particularly valuable for ranking compounds across multiple parameters and identifying those with the highest potential for further development.
Desirability Functions Desirability functions transform individual property values into dimensionless desirability scores (typically 0-1 range), which are then combined into an overall composite metric [123]. This approach enables systematic comparison of compounds across diverse parameter scales and units. For PARP-1 inhibitors, specific desirability functions have been identified that characterize approved drugs, providing valuable benchmarks for designing better candidates [123].
Pareto Optimization Pareto optimization identifies compounds where improvement in one parameter necessitates deterioration in another, creating an "optimal frontier" of candidate compounds [121]. This approach is particularly valuable for visualizing and navigating trade-offs between conflicting parameters such as lipophilicity and solubility. Advanced implementations like the STELLA framework demonstrate efficient Pareto front advancement in complex multi-parameter spaces [124].
AI and Deep Learning Approaches Modern deep learning-based de novo design methods can generate novel small molecules while optimizing multiple parameters simultaneously, including late-stage development parameters [125] [124]. These approaches leverage generative models and multi-objective reinforcement learning to explore chemical space more efficiently than traditional methods. The STELLA framework, for example, combines evolutionary algorithms with deep learning models to achieve extensive fragment-level chemical space exploration while balancing multiple parameters [124].
Experimental Protocols and Methodologies
Comprehensive Lipophilicity and ADMET Profiling Workflow A robust MPO strategy integrates both computational predictions and experimental validation throughout the drug discovery process:
Lipophilicity-ADMET Profiling Workflow
Detailed RP-TLC Protocol for Lipophilicity Determination Reversed-phase thin-layer chromatography provides a robust, high-throughput method for experimental lipophilicity assessment [11]:
- Stationary Phase: RP-18 F254s TLC plates
- Mobile Phase: Acetone-TRIS buffer (pH 7.4) mixtures, varying proportions
- Sample Application: 0.5-1 μL of 1 mM compound solutions in methanol
- Chromatography Chamber: Saturated with mobile phase vapor, 20 ± 1°C
- Detection: UV light at 254 nm and 365 nm
- R₀ Determination: Extrapolated to zero organic modifier concentration
- log P Calculation: From correlation equation log P = aR₀ + b
This method was successfully applied to diquinothiazine hybrids, demonstrating strong correlation with computational predictions and providing reliable experimental lipophilicity data for MPO decision-making [11].
In Silico ADMET Profiling Protocols Computational ADMET prediction platforms enable rapid screening of compound libraries for key properties:
- SwissADME: Provides free access to multiple prediction models for gastrointestinal absorption, blood-brain barrier penetration, CYP450 inhibition, and other critical parameters [11]
- pkCSM: Offers predictive models for permeability, distribution, metabolism, and toxicity endpoints, serving as a complementary tool to SwissADME [11]
- StarDrop: Incorporates ADMET QSAR models with probabilistic scoring for MPO, including P-gp transport, human intestinal absorption, and hERG inhibition [122]
These platforms generate predictive data that can be integrated into MPO frameworks, though predictions should be validated experimentally for key compounds.
Case Studies: Successful MPO Implementation
Case Study 1: Diquinothiazine Hybrids as Anticancer Agents A comprehensive MPO approach was applied to novel dialkylaminoalkyldiquinothiazine hybrids with demonstrated anticancer activity [11]. Researchers integrated experimental RP-TLC lipophilicity measurements with computational predictions using eight different log P algorithms. The study found that chromatographically determined lipophilicity parameters (R₀) showed the biggest similarity with iLOGP predictions among the tested algorithms. This combined experimental-computational approach enabled efficient optimization of lipophilicity alongside anticancer potency, resulting in compounds with IC₅₀ values below 3 μM against various cancer cell lines while maintaining favorable ADMET profiles [11].
Case Study 2: PARP-1 Inhibitor Optimization MPO strategies for PARP-1 inhibitors have identified specific desirability functions based on approved drugs' physicochemical and pharmacokinetic properties [123]. Researchers analyzed the property space of successful PARP-1 inhibitors to establish target ranges for lipophilicity, molecular weight, polar surface area, and other key parameters. This analysis informed the design of novel inhibitors with improved selectivity and reduced toxicity profiles. The integration of matched molecular pair analysis (MMPA) further identified statistically meaningful structural transformations that enhance activities while maintaining optimal lipophilicity [123].
Case Study 3: STELLA Framework for Multi-Parameter Optimization The STELLA framework demonstrates advanced MPO capabilities through its combination of evolutionary algorithms with deep learning-based property prediction [124]. In a comparative study focusing on PDK1 inhibitors, STELLA generated 217% more hit candidates with 161% more unique scaffolds compared to REINVENT 4, while achieving better average scores for both docking (GOLD PLP Fitness: 76.80 vs. 73.37) and drug-likeness (QED: 0.78 vs. 0.75) [124]. The framework's clustering-based conformational space annealing method effectively balances exploration of diverse chemical space with optimization of multiple parameters, including lipophilicity-dependent properties.
Research Reagents and Computational Tools
Table 3: Essential Research Reagents and Computational Tools for MPO
| Category | Tool/Reagent | Specific Function | Application in MPO |
|---|---|---|---|
| Computational Platforms | SwissADME | Free web tool for ADMET prediction | Rapid screening of absorption, distribution, and metabolism parameters [11] |
| pkCSM | Predictive pharmacokinetic and toxicity platform | Complementary ADMET profiling to SwissADME [11] | |
| StarDrop | Integrated drug discovery platform with MPO scoring | Probabilistic scoring combining multiple parameters with uncertainty estimates [122] | |
| STELLA | Metaheuristics-based generative molecular design | Fragment-based chemical space exploration with multi-parameter optimization [124] | |
| Experimental Assays | RP-TLC System | Chromatographic lipophilicity determination | Experimental R₀ measurement for correlation with computed log P values [11] |
| Caco-2 Cell Assays | Intestinal permeability assessment | Prediction of oral absorption potential [122] | |
| MDCK-MDR1 | Transporter-mediated efflux evaluation | P-glycoprotein interaction profiling [122] | |
| Liver Microsomes | Metabolic stability assessment | Phase I metabolism evaluation [122] | |
| Specialized Reagents | P-gp Membranes | Isolated membrane fractions expressing P-glycoprotein | Transporter inhibition screening [122] |
| CYP450 Isozymes | Individual cytochrome P450 enzymes | Metabolic pathway identification and drug-drug interaction potential [122] | |
| hERG Assay Kits | Potassium channel binding tests | Cardiotoxicity risk assessment [122] |
Multi-parameter optimization represents an essential strategy for modern drug discovery, with lipophilicity serving as a central parameter due to its profound influence on ADMET properties. Successful MPO requires integrated approaches that combine computational predictions with experimental validation, leveraging diverse methodologies including probabilistic scoring, desirability functions, Pareto optimization, and AI-driven generative design. The case studies presented demonstrate that systematic MPO implementation can yield compounds with balanced properties, reducing development attrition and increasing the probability of clinical success.
Future directions in MPO will likely involve increased incorporation of explainable AI methods that not only optimize parameters but also provide structural insights into the molecular features driving property relationships [125]. Additionally, the integration of advanced metaheuristic algorithms with deep learning models, as demonstrated by frameworks like STELLA, points toward more efficient navigation of chemical space while balancing complex, often conflicting parameters [124]. As these methodologies continue to evolve, MPO will remain indispensable for addressing the fundamental challenge of designing drugs that are not only potent but also developable.
Addressing Challenges Specific to Protein-Protein Interaction Inhibitors
Protein-protein interactions (PPIs) govern fundamental cellular processes, making them attractive therapeutic targets for various diseases, including cancer, neurodegenerative disorders, and infectious diseases [126] [127]. However, the classical "small molecule" approach to drug discovery has largely overlooked PPIs in favor of targeting individual proteins like enzymes and receptors [126]. This historical neglect stems from the significant challenges inherent in modulating PPIs, primarily due to their characteristically large, flat, and hydrophobic interfaces, which lack the deep binding pockets typical of conventional drug targets [126] [127]. Despite these hurdles, the field has matured, with several PPI modulators now receiving clinical approval, demonstrating that targeting PPIs is a viable and promising strategy for drug development [126] [127]. A critical factor in the success of any small-molecule therapeutic, especially PPI inhibitors, is its pharmacokinetic profile, which is profoundly influenced by physicochemical properties like lipophilicity. This guide examines the core challenges in developing PPI inhibitors and outlines advanced strategies to overcome them, with a particular focus on the critical impact of lipophilicity on Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties.
Fundamental Challenges in PPI Inhibitor Development
Structural and Energetic Characteristics of PPI Interfaces
The biological and structural nature of PPI interfaces presents the first and most significant set of obstacles:
- Large and Flat Surfaces: Unlike the deep, concave binding pockets of enzymes, PPI interfaces are typically extensive (ranging from 1,500-3,000 Ų) and remarkably flat, offering few grooves or cavities for small molecules to bind with high affinity [126] [127].
- Hydrophobicity: The interfaces are often highly hydrophobic, which, while offering a potential driving force for binding, can lead to compounds with poor aqueous solubility and increased promiscuity [126].
- High-Affinity Residue Binding: The binding energy is often distributed across a wide area, with a handful of key residues, known as "hot spots," contributing disproportionately to the binding free energy. Displacing these high-affinity interactions with a small molecule is energetically challenging [126] [127].
The Critical Role of Lipophilicity in PPI Inhibitor ADMET Profiles
The very properties that enable a molecule to engage a hydrophobic PPI interface often create unfavorable ADMET profiles. Lipophilicity, quantified as the partition coefficient P (or its logarithm, logP) between octanol and water, is a principal parameter determining a drug's pharmacokinetic behavior [15] [128]. Its impact is multifaceted:
- Absorption and Permeability: Compounds with moderate lipophilicity are generally better absorbed through cell membranes. However, PPI inhibitors often require high molecular weight and lipophilicity to achieve sufficient binding, which can compromise oral bioavailability [15].
- Distribution: Highly lipophilic compounds can readily cross membranes but may sequester in lipid-rich tissues, leading to a large volume of distribution and potential accumulation [15].
- Metabolism and Toxicity: Increased lipophilicity often correlates with higher metabolic clearance and a greater risk of off-target toxicity and promiscuous binding [15] [128]. This is a particular concern for PPI inhibitors, as their physicochemical starting point is already suboptimal.
The following table summarizes the key challenges and their implications for drug discovery.
Table 1: Core Challenges in Developing Small-Molecule PPI Inhibitors
| Challenge | Description | Consequence for Drug Discovery |
|---|---|---|
| Interface Topography | Large (1,500-3,000 Ų), flat, and relatively featureless binding surfaces [126] [127]. | Poor complementarity for small molecules; difficult to achieve sufficient binding affinity. |
| "Hot Spot" Dependency | Binding energy is non-uniformly distributed across a few critical residues [126] [127]. | Requires precise targeting of specific regions; traditional HTS may be ineffective. |
| High Lipophilicity | Compounds often need high logP to engage hydrophobic interfaces [15]. | Poor solubility, high metabolic clearance, increased risk of toxicity, and promiscuity. |
| Ligand Efficiency | High molecular weight often needed for sufficient binding energy at large interface [126]. | Poor pharmacokinetics and increased likelihood of violating drug-likeness rules (e.g., Lipinski's Rule of 5). |
Strategic Approaches to PPI Inhibitor Discovery
Experimental and Computational Screening Methodologies
Given the limitations of traditional high-throughput screening (HTS) against PPIs, several specialized strategies have been developed:
- Fragment-Based Drug Discovery (FBDD): This approach uses small, low molecular weight fragments (<250 Da) that bind weakly to distinct sub-pockets on the PPI interface [126] [127]. Fragment libraries are particularly useful for targeting flatter surfaces without well-defined pockets. The binding of a fragment reveals a new chemical space that can be grown or linked to other fragments to develop a high-affinity drug lead [126].
- Structure-Based Virtual Screening: This computational method leverages 3D structural information of the target protein to screen large virtual compound libraries in silico [127]. A successful example is the identification of stabilizers for the 14-3-3/ChREBP interaction by performing molecular docking focused on the phospho-accepting pocket at the PPI interface [129].
- Targeting Allosteric Sites: Instead of competing directly with the protein partner at the orthosteric site, allosteric inhibitors bind to a topologically distal site, inducing a conformational change that disrupts the PPI [126]. Allosteric pockets can be more structured and druggable than the primary interface, offering improved physiochemical and pharmacological properties for the inhibitors [126].
The "Scientist's Toolkit": Key Reagents and Methods
A multidisciplinary toolkit is essential for modern PPI inhibitor discovery. The table below lists key reagents, computational tools, and experimental methods cited in recent research.
Table 2: Research Reagent Solutions for PPI Inhibitor Development
| Tool / Reagent / Method | Function / Application | Example Use Case |
|---|---|---|
| Fragment Libraries | Identify low molecular weight binders to "hot spot" regions on flat PPI interfaces [126]. | Initial hit finding in FBDD campaigns against challenging PPI targets. |
| SwissADME / pkCSM | Online platforms for predicting ADMET parameters and drug-likeness [15] [128]. | Early-stage in silico profiling of absorption, distribution, and toxicity of hit/lead compounds. |
| RP-TLC (Reverse Phase Thin-Layer Chromatography) | Experimental determination of lipophilicity (expressed as RM0 or logPTLC) [15] [128]. | Measuring compound lipophilicity during lead optimization; requires smaller sample amounts than shake-flask. |
| Dibenzo[b,e]azepin-6(6H)-one Scaffold | A tricyclic chemical scaffold used as a core structure for designing PPI inhibitors [130] [131]. | Served as a starting point for designing inhibitors of the PEX5-PEX14 PPI in Trypanosoma [130]. |
| AlphaFold / RosettaFold | AI-based protein structure prediction tools [132] [127]. | Generating 3D structural models of PPI targets and complexes for structure-based design when experimental structures are unavailable. |
Lipophilicity and ADMET Profiling: Methods and Data
Experimental and Computational Determination of Lipophilicity
Accurate assessment of lipophilicity is non-negotiable. The following experimental and computational approaches are standard:
- Chromatographic Methods (RP-TLC): Reverse-phase Thin-Layer Chromatography is a widely used, robust, and low-sample-requirement method for determining lipophilicity [15] [128]. The chromatographic parameter RM0 is determined and can be converted to an experimental logP value (logPTLC). This method is advantageous over the traditional shake-flask technique due to its speed and ability to handle impure samples [15].
- Computational Predictions: Numerous in silico algorithms exist for predicting logP, including iLOGP, XLOGP3, WLOGP, MLOGP, and SILCOS-IT [15] [128]. These are classified into atom-based, fragment-based, topology-based, and structural-property-based methods, each with strengths and weaknesses. For instance, a study on diquinothiazines found the iLOGP algorithm showed the biggest similarity to the experimental RM0 values for certain compounds [15].
Quantitative Lipophilicity and ADMET Data from Recent Studies
Recent studies provide concrete data linking lipophilicity to biological activity and ADMET parameters. The table below synthesizes key quantitative findings from the literature.
Table 3: Experimental Lipophilicity and ADMET Data of Investigational Compounds
| Compound Class / Study | Experimental logP (Method) | Computational logP (Algorithm) | Key ADMET & Bioactivity Findings |
|---|---|---|---|
| Diquinothiazine Hybrids [15] | RM0 values determined (RP-TLC) | iLOGP, XLOGP3, WLOGP, MLOGP, SILCOS-IT | Compounds with high activity (IC50 < 3 μM) against lung cancer (A549) and glioblastoma (SNB-19) cell lines were identified. In silico tools predicted favorable pharmacokinetic profiles. |
| Tetracyclic Chlorpromazine Analogues [128] | logPTLC determined (RP-TLC) | iLOGP, XLOGP3, WLOGP, MLOGP, SILCOS-IT, ClogP | The tested compounds demonstrated promising activity against A549 lung cancer cells without affecting healthy HaCaT cells. SwissADME and pkCSM predicted favorable ADME parameters. |
| 14-3-3/ChREBP Stabilizer (Compound 3) [129] | N/R | N/R | Structure-based optimization yielded a selective stabilizer (EC50 5.2 μM) that enhanced the PPI by 14-fold. The crystal structure revealed a "molecular glue" binding mode. |
Abbreviations: N/R = Not Reported in the cited source; RP-TLC = Reverse Phase Thin-Layer Chromatography.
Detailed Experimental Protocols
Protocol for Determining Lipophilicity Using RP-TLC
This protocol is adapted from methodologies described in the search results [15] [128].
- Plate Preparation: Use commercially available TLC plates pre-coated with silica gel RP-18 F254.
- Sample Application: Spot solutions of the test compounds (and standards if used) approximately 1.5 cm from the bottom edge of the plate.
- Mobile Phase Preparation: Prepare a series of mobile phases consisting of a water-miscible organic modifier (e.g., acetone or methanol) and an aqueous TRIS buffer (pH 7.4). The volume fraction of the organic modifier should range from, for example, 50% to 80% (v/v) in 5-10% increments.
- Chromatogram Development: Develop the chromatograms in a chromatographic chamber saturated with the mobile phase vapor. The development distance is typically 8-10 cm.
- Detection: Detect the spots under UV light (at 254 nm) or using an appropriate visualizing agent.
- Data Calculation:
- Calculate the retention factor RF for each compound: RF = distance traveled by compound / distance traveled by solvent front.
- Calculate the RM value: RM = log (1/RF - 1).
- For each compound, plot RM values against the volume fraction of the organic modifier in the mobile phase. The lipophilicity parameter RM0 is the intercept value on the RM axis (extrapolated to 0% organic modifier). This RM0 value can be used as a direct measure of lipophilicity or converted to logPTLC.
Protocol for a Structure-Based Virtual Screening Cascade
This protocol is based on the successful identification of 14-3-3 PPI stabilizers [129].
- Target Preparation: Obtain or generate a high-resolution 3D structure of the PPI complex. Identify a potential binding pocket at the interface (e.g., a sub-pocket adjacent to a known "hot spot").
- Compound Library Preparation: Curate a large, commercially available virtual compound library (e.g., MolPort, ZINC). Prepare the structures by adding hydrogens, generating plausible tautomers and protonation states at physiological pH, and performing energy minimization.
- Structure-Based Filtering: Apply substructure or pharmacophore filters to narrow the library focus. For example, a screen for 14-3-3 binders filtered for compounds containing a phosphate or phosphonate group [129].
- Molecular Docking: Dock the filtered library into the defined binding pocket using a standard docking program (e.g., Glide). Rank the compounds based on their docking scores.
- Induced Fit Docking (IFD): Subject the top-ranked hits to an IFD protocol, which allows for side-chain and backbone flexibility in the binding site, to refine the poses and account for protein conformational changes upon ligand binding.
- Visual Inspection and Selection: Manually inspect the top IFD poses for sensible binding modes, key interactions (e.g., hydrogen bonds, hydrophobic contacts), and chemical attractiveness. Select a final set of compounds for purchase and experimental validation.
The following workflow diagram illustrates the key steps and decision points in this process.
The development of potent and selective protein-protein interaction inhibitors remains a formidable challenge in drug discovery. Success requires an integrated strategy that combines innovative discovery methods like FBDD and structure-based virtual screening with a relentless focus on optimizing ADMET properties from the earliest stages. As demonstrated, lipophilicity is a central parameter that sits at the crossroads of achieving target binding and ensuring a viable pharmacokinetic profile. Mastering the interplay between molecular structure, lipophilicity, and ADMET outcomes through continuous experimental and computational profiling is paramount. The strategies and protocols outlined in this guide provide a roadmap for researchers to navigate the complex landscape of PPI inhibitor development, ultimately increasing the probability of translating promising chemical matter into effective therapeutics.
Validation and Comparative Analysis of Lipophilicity-Guided Strategies
Benchmarking Computational Predictions Against Experimental Data
Lipophilicity, the measure of how readily a compound dissolves in nonpolar solvents versus water, is a principal parameter governing the pharmacokinetic behavior of a drug candidate. It critically influences a compound's Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET), with deficiencies in these properties accounting for a significant proportion of failures in clinical trials [15] [133]. In the context of modern drug discovery, computational predictions of lipophilicity and other ADMET properties offer a high-throughput, low-cost alternative to experimental assays. However, the reliability of these in silico tools is not absolute, necessitating a rigorous process of benchmarking—the systematic evaluation of computational predictors against robust experimental datasets—to assess their predictive accuracy, limitations, and applicability to novel chemical spaces [133] [134]. This guide details the methodologies and protocols for the effective benchmarking of computational lipophilicity predictions within ADMET research.
Computational Prediction Landscape
A wide array of software tools implementing Quantitative Structure-Activity Relationship (QSAR) models are available for predicting lipophilicity and other physicochemical properties. Benchmarking studies are essential for identifying the most robust tools among them [133].
The selection of software for benchmarking should include a mix of freely available and commercial tools, prioritizing those capable of batch predictions and providing applicability domain (AD) assessments [133]. Key tools identified in recent benchmarks include:
- OPERA (Open (Quantitative) Structure–activity/property Relationship App): An open-source battery of QSAR models from the U.S. National Institute of Environmental Health Science (NIEHS) for predicting various physicochemical properties [133].
- SwissADME: A widely used web tool that provides predictions for a range of pharmacokinetic properties, including lipophilicity via several calculated logP methods [15].
- pkCSM: A platform used for predicting a wide spectrum of ADMET parameters, allowing for correlation with experimental lipophilicity data [15].
Performance Metrics and Benchmarking Findings
The predictive performance of computational models is quantitatively assessed using standardized metrics. For regression tasks like logP prediction, common metrics include the Root Mean Squared Error (RMSE) and the coefficient of determination (R²). For instance, a study on peptide lipophilicity reported an RMSE of 0.47 for a support vector regression model on an external test set [135]. Overall, models predicting physicochemical properties tend to show strong performance, with benchmarks reporting an average R² of 0.717 [133].
Table 1: Common Metrics for Benchmarking Regression and Classification Models
| Task Type | Key Metrics | Interpretation |
|---|---|---|
| Regression (e.g., logP) | Root Mean Squared Error (RMSE) | Lower values indicate higher accuracy. |
| Mean Absolute Error (MAE) | Average magnitude of errors, less sensitive to outliers. | |
| Coefficient of Determination (R²) | Proportion of variance explained by the model; closer to 1 is better. | |
| Classification (e.g., Toxicity) | Area Under the ROC Curve (AUROC) | Measures the ability to distinguish between classes; 1 is perfect. |
| Area Under the Precision-Recall Curve (AUPRC) | Suitable for imbalanced datasets. | |
| Matthews Correlation Coefficient (MCC) | A balanced measure for binary classifications. |
Recent benchmarking efforts reveal that classical machine learning methods, such as random forests and gradient-boosted trees using fixed molecular fingerprints (e.g., ECFP), remain highly competitive for many ADMET tasks [134]. However, more advanced methods like Graph Neural Networks (GNNs), which learn features directly from the molecular graph, have demonstrated superior generalization, particularly in challenging out-of-distribution (OOD) scenarios [134].
Experimental Data as the Gold Standard
Computational predictions must be validated against reliable experimental data, which serves as the benchmark for evaluating predictive accuracy.
Experimental Methods for Determining Lipophilicity
Two primary experimental approaches are used to determine lipophilicity, quantified as the partition coefficient (P) or its decimal logarithm (logP), and the distribution coefficient (logD) at a specific pH [15].
- Shake-Flask Method: This is the classical gold standard method, involving the direct measurement of a compound's concentration between two immiscible phases, typically 1-octanol and water. While accurate for logP values in the range of -2 to 4, it is time-consuming, requires relatively large amounts of pure compound, and demands careful control of experimental parameters [15].
- Chromatographic Techniques: Reversed-phase chromatographic methods, such as Reversed-Phase Thin-Layer Chromatography (RP-TLC) and Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC), are the most widely used indirect methods.
- RP-TLC Methodology: The compound is applied to a reversed-phase (e.g., RP-18) plate and developed with a mobile phase consisting of a water-miscible organic modifier (e.g., acetone) and an aqueous buffer (e.g., TRIS buffer at pH 7.4). The lipophilicity parameter (R₀) is derived from the relationship between the compound's retention factor (R₍M₎) and the concentration of the organic modifier [15].
- Advantages: Chromatographic methods require smaller sample amounts, are faster, and provide highly repeatable results that correlate well with shake-flask values (typically within ±1 log unit) [15].
Curating Experimental Benchmark Datasets
The quality of the experimental dataset is paramount for a meaningful benchmark. Data must be collected from literature sources and rigorously curated [133] [134].
- Data Collection: Sources like ChEMBL and the Therapeutics Data Commons (TDC) provide experimental data. Search terms must be exhaustive and include standard abbreviations for the endpoints of interest [133].
- Data Curation Workflow: A standardized curation procedure is critical. This includes:
- Standardization: Converting chemical structures to standardized SMILES notation.
- Removal of Inorganics/Organometallics: Filtering out compounds with unusual elements.
- Neutralization of Salts: Ensuring the neutral form of the compound is considered.
- Handling Duplicates: Averaging values for duplicates with small deviations and removing those with high variability.
- Outlier Removal: Identifying and removing intra-dataset and inter-dataset outliers using statistical methods like Z-score analysis [133].
Data Curation Workflow
A Framework for Benchmarking
A robust benchmarking framework goes beyond a simple random split of data, aiming to evaluate how models perform on chemically novel compounds.
Dataset Partitioning Strategies
To simulate real-world scenarios where models predict properties for entirely new chemical scaffolds, benchmarks should employ several data-splitting strategies [134]:
- Random Splitting: The dataset is randomly divided into training and test sets. This provides a baseline performance measure but can lead to over-optimistic results due to chemical similarity between sets.
- Scaffold Splitting: Molecules are divided based on their core molecular framework (scaffold). This tests a model's ability to extrapolate to structurally novel compounds, which is a more realistic assessment of its utility in drug discovery [134].
- Temporal Splitting: Data is split based on the date of publication or testing. This evaluates performance on compounds that were discovered or synthesized after the model was trained, mimicking a real-world temporal validation [134].
Evaluating Model Performance and Applicability Domain
The core of benchmarking involves running predictions on the test set and comparing them to experimental values using the metrics in Table 1. A critical, often overlooked, aspect is the Applicability Domain (AD)—the chemical space within which the model makes reliable predictions. Predictions for compounds outside the model's AD should be treated with caution. Tools that provide AD assessment (e.g., via leverage or similarity measures) are highly valuable [133].
Table 2: Key Reagents and Materials for Experimental Lipophilicity Determination
| Reagent/Material | Function in Experiment | Example Specification |
|---|---|---|
| 1-Octanol | Organic solvent in the shake-flask method to simulate the lipid membrane environment. | HPLC grade, >99% purity [15]. |
| TRIS Buffer (pH 7.4) | Aqueous phase in RP-TLC; maintains physiological pH for logD7.4 measurement. | 0.05 M concentration, pH adjusted with HCl [15]. |
| RP-18 TLC Plates | Stationary phase in Reversed-Phase Thin-Layer Chromatography. | Commercial silica plates with bonded C18 chains [15]. |
| Acetone | Organic modifier in the mobile phase for RP-TLC, modulates compound retention. | HPLC grade [15]. |
| UV/Visualization Kit | For detecting compounds on TLC plates after development. | UV lamp at 254 nm or chemical staining reagents. |
Integrated Benchmarking Workflow
The following workflow integrates the concepts and methodologies detailed in the previous sections into a practical, step-by-step guide for researchers.
Benchmarking Workflow
Benchmarking computational predictions against experimental data is a cornerstone of reliable ADMET research. As demonstrated, a rigorous approach involves careful tool selection, meticulous experimental data curation, and the use of chemically meaningful validation protocols like scaffold splitting. The field is advancing with the incorporation of more sophisticated models like Graph Neural Networks and a heightened focus on out-of-distribution robustness. By adhering to a structured benchmarking framework, researchers can confidently identify the best in silico tools, thereby de-risking the drug discovery pipeline and accelerating the development of safer, more effective therapeutics.
Lipophilicity, quantified by the octanol-water partition coefficient (LogP), is a fundamental physicochemical property that profoundly influences the absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles of therapeutic substances [136] [137]. It serves as a critical determinant in a drug's behavior, governing its solubility, passive membrane permeability, and overall bioavailability [137]. In the early stages of drug design and development, the rapid and accurate prediction of LogP is therefore indispensable for prioritizing candidate molecules with optimal pharmacokinetic and pharmacodynamic profiles [136] [138].
The partition coefficient, LogP, is a constant for a given compound and measures the distribution of its neutral (unionized) form between octanol and water [2]. In contrast, the distribution coefficient, LogD, is pH-dependent and accounts for the distribution of all forms of the compound—ionized, partially ionized, and unionized—at a specific pH, making it a more accurate descriptor of lipophilicity under physiological conditions [2]. Despite the importance of LogD, the accurate calculation of LogP remains the foundational step upon which many LogD models are built.
This review provides a comparative analysis of contemporary in silico LogP prediction algorithms, assessing their methodological foundations, relative predictive accuracies, and utility within the context of modern ADMET research.
Computational methods for predicting LogP can be broadly classified into several categories based on their fundamental approach to correlating molecular structure with lipophilicity. The following table summarizes the primary types of algorithms and their underlying principles.
Table 1: Classification of LogP Prediction Methods
| Method Type | Underlying Principle | Representative Algorithms | Key Characteristics |
|---|---|---|---|
| Substructure-Based | Summation of contributions from molecular fragments or individual atoms [138]. | CLOGP, XLOGP2, WLOGP [139] | Relies on predefined fragment libraries; performance depends on fragment coverage. |
| Property-Based | Utilizes molecular descriptors or whole-molecule properties, often derived from 3D structure or topology [138]. | iLOGP, MLOGP, ALOGPS [139] | Includes approaches using topological descriptors and molecular simulation. |
| Similarity Search | Compares the query molecule to a database of compounds with known LogP values, using the experimental value of the most similar compound as a starting point [139]. | XLOGP3 [139] | Accuracy scales with the degree of similarity to known compounds in the database. |
A novel approach, MF-LOGP, demonstrates that it is possible to predict LogP using only molecular formula as input, without any structural information. This random forest algorithm uses features derived from the molecular formula, such as atom counts, and has been shown to achieve performance competitive with some structure-based methods (R² = 0.83, RMSE = 0.77) [139]. This is particularly useful for complex mixtures where structural data is unavailable.
Comparative Performance Analysis of Key Algorithms
Evaluations on large, diverse datasets are essential for understanding the real-world applicability of these prediction tools. A comprehensive study comparing 30 methods on a public dataset and 18 methods on two large industrial datasets (including one with over 95,000 compounds from Pfizer) revealed that the accuracy of most models declines as the number of non-hydrogen atoms in a molecule increases [138]. Strikingly, the study proposed a simple, highly effective equation based solely on the number of carbon (NC) and heteroatoms (NHET): LogP = 1.46 + 0.11 NC - 0.11 NHET, which outperformed a significant number of the benchmarked programs [138].
The performance of various algorithms, as reported in independent analyses, is summarized in the table below.
Table 2: Comparative Performance of Selected LogP Prediction Algorithms
| Algorithm | Reported RMSE (Log Units) | Method Category | Key Findings from Literature |
|---|---|---|---|
| ALOGPS | 1.02 [139] | Property-Based (Neural Network) | Noted for its high accuracy in independent studies [139]. |
| XLOGP3 | 1.08 [139] | Similarity Search | Introduction of a similarity search improved the accuracy of its predecessor (XLOGP2) by 40% [139]. |
| CLOGP | 1.23 [139] | Substructure-Based (Fragmental) | An early pioneer with an extensive and updated fragment library [139]. |
| MLOGP | 2.03 [139] | Property-Based (Linear Regression) | Simpler model, showing higher error compared to more complex models [139]. |
| MF-LOGP | 0.77 [139] | Formula-Based (Random Forest) | Competitive accuracy using only molecular formula, no structural input. |
| iLOGP | Information Missing | Property-Based (Molecular Simulation) | Uses 3D structure to approximate electron densities and molecular size [139]. |
Only seven of the eighteen methods tested on the large industrial datasets were deemed successful, highlighting the challenge of achieving consistent accuracy across a broad drug-like chemical space [138]. Factors such as the presence of ionizable groups, complex stereochemistry, and uncommon structural motifs can significantly impact prediction reliability.
The Critical Link Between LogP and ADMET Properties
Lipophilicity is deeply intertwined with a compound's ADMET profile, influencing multiple key processes [136] [137].
- Absorption and Permeability: Optimal LogP is crucial for good intestinal absorption via passive diffusion. According to Lipinski's Rule of Five, an orally active drug should typically have a LogP value less than 5 [137]. For drugs targeting the central nervous system, which must cross the blood-brain barrier, the ideal LogP is around 2 [137]. The topological polar surface area (TPSA), often calculated alongside LogP, is another strong indicator of a compound's ability to penetrate the blood-brain barrier [136].
- Metabolism and Toxicity: Highly lipophilic compounds are more prone to nonspecific binding and sequestration in fatty tissues, which can lead to increased systemic toxicity and slow excretion [137]. Furthermore, lipophilicity is a key parameter in comprehensive ADMET scoring functions, such as the ADMET-score, which integrates predictions from 18 different ADMET endpoints—including CYP450 enzyme inhibition, human intestinal absorption, and hERG channel inhibition—to provide a unified metric for evaluating drug-likeness [100].
- Distribution and Efflux: Lipophilicity can influence a compound's interaction with efflux transporters like P-glycoprotein (P-gp). P-gp is a critical determinant of drug distribution and elimination, and its substrates often exhibit specific lipophilicity ranges [140]. Assessing the DDI risk of P-gp substrates involves a combination of in vitro efflux ratios and apparent permeability coefficients (Papp), parameters that are indirectly related to compound lipophilicity [141].
Experimental Validation and Research Workflows
Key Reagents and Experimental Tools
Experimental validation remains the gold standard for lipophilicity assessment. The following table outlines key materials and methods used in experimental protocols cited in the literature.
Table 3: Essential Research Reagents and Methods for Lipophilicity Assessment
| Reagent / Material | Function in Experiment | Example Use Case |
|---|---|---|
| RP-TLC Plates (e.g., RP-18F₂₅₄) | Stationary phase for Reverse-Phase Thin-Layer Chromatography, mimicking the lipophilic environment [136]. | Experimental determination of the lipophilicity parameter (RMW) for neuroleptics [136]. |
| Organic Modifiers (Acetone, Acetonitrile, 1,4-dioxane) | Components of the mobile phase in RP-TLC, used to create gradients for elution [136]. | Used in combination with different stationary phases to find optimal chromatographic conditions [136]. |
| P-gp Expressing Cell Lines (e.g., Caco-2, LLC-PK1) | In vitro model to study active efflux and permeability, processes influenced by lipophilicity [141]. | Assessment of efflux ratio and apparent permeability (Papp) for DDI risk prediction [141]. |
| Mdr1a/1b Knockout Mice | In vivo model to study the systemic impact of P-gp-mediated efflux on pharmacokinetics [141]. | Used to correlate in vitro P-gp substrate data with in vivo exposure (AUC) [141]. |
Workflow for Integrated Lipophilicity Assessment in Drug Discovery
A robust approach to lipophilicity assessment in drug discovery integrates both computational and experimental techniques. The following diagram illustrates a proposed workflow for this integrated assessment, highlighting the role of LogP prediction at various stages.
Protocol for Reverse-Phase TLC (RP-TLC) Lipophilicity Determination
As detailed in the research on neuroleptics, a standard protocol for experimental lipophilicity determination via RP-TLC involves the following steps [136]:
- Stationary Phase Selection: Use reverse-phase TLC plates, such as RP-2F254, RP-8F254, or RP-18F254, which provide different levels of hydrophobicity.
- Mobile Phase Preparation: Prepare mobile phases consisting of binary mixtures involving water and an organic modifier. Common modifiers include acetone, acetonitrile, or 1,4-dioxane. A series of mobile phases with varying volume fractions of the organic modifier are typically used.
- Chromatography Development: Spot the compounds of interest on the TLC plate and develop the chromatogram in a pre-saturated chamber.
- Retention Factor (RM) Calculation: After development, measure the retention factor (Rf) for each compound and calculate the RM value using the formula: RM = log(1/Rf - 1).
- Lipophilicity Index (RMW) Determination: The RM values are plotted against the concentration of the organic modifier. The lipophilicity index (RMW) is extrapolated from the linear relationship as the theoretical RM value in pure water, which can be interpreted as an experimental logP value [136].
The accurate prediction of LogP remains a dynamic and critical area of research in cheminformatics and drug discovery. While a multitude of algorithms exist—ranging from fragment-based and property-based to novel AI-driven approaches—their performance varies significantly across chemical space. No single algorithm universally outperforms all others, underscoring the need for researchers to select methods based on the specific chemical series under investigation.
Future advancements will likely stem from the curation of larger, higher-quality experimental datasets, such as those in PharmaBench, and the intelligent integration of multiple prediction methods, perhaps through consensus models or advanced machine learning techniques that can better capture complex molecular interactions [20]. As the field moves further beyond the Rule of Five to explore larger and more complex molecules, the development of robust predictors that can accurately estimate the lipophilicity of these challenging compounds will be paramount to reducing late-stage attrition in drug development.
The optimization of absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties represents a critical hurdle in modern drug discovery, with poor pharmacokinetic profiles and safety concerns remaining leading causes of clinical-stage attrition [81]. Among the fundamental physicochemical parameters influencing ADMET behavior, lipophilicity serves as a principal determinant that governs a compound's interaction with biological systems [15].
This whitepaper presents a detailed technical analysis of successful drug candidate optimization through the lens of lipophilicity management, featuring a contemporary case study on anticancer diquinothiazines. We provide comprehensive experimental methodologies, quantitative structure-property relationship analyses, and visualization of the strategic workflows that enable researchers to navigate the complex interplay between molecular structure, lipophilicity, and pharmacokinetic outcomes.
The Central Role of Lipophilicity in ADMET Optimization
Lipophilicity, quantitatively expressed as the partition coefficient (log P) or distribution coefficient (log D), directly influences virtually all ADMET parameters [15]. Compounds with moderate lipophilicity typically demonstrate superior absorption profiles due to enhanced passive permeability across cellular membranes. Furthermore, distribution characteristics, including tissue penetration and volume of distribution, correlate strongly with lipophilic character [81].
Excessively lipophilic compounds often present significant development challenges, including:
- Poor aqueous solubility limiting oral bioavailability
- Increased metabolic clearance through cytochrome P450 pathways
- Higher risk of promiscuous binding and toxicity
- Accumulation in lipid-rich tissues leading to prolonged elimination half-lives [15]
The optimal lipophilicity range for successful drug candidates typically falls between log P 1-3, balancing membrane permeability with sufficient aqueous solubility for dissolution and absorption [81]. Recent advances in machine learning (ML) and artificial intelligence (AI) have enhanced our ability to predict these relationships, with AI-discovered molecules now demonstrating 80-90% success rates in Phase I clinical trials, substantially higher than historical industry averages [142].
Case Study: Anticancer Diquinothiazines with Optimized Pharmacophore Substituents
Compound Series and Biological Activity Profile
A recent investigation of fifteen newly synthesized dialkylaminoalkyldiquinothiazine hybrids provides a compelling case study in systematic lipophilicity optimization [15]. These angularly condensed diquinothiazines were designed with specific pharmacophore substituents to enhance their anticancer potency while maintaining favorable ADMET properties.
The compounds were evaluated against multiple cancer cell lines, with the most promising candidates demonstrating exceptional activity:
- Compound 2 (dimethylaminopropyldiquinothiazine): IC₅₀ = 0.3 μM against A549 lung cancer cells
- Compound 8 (pyrrolidinylethyldiquinothiazine): IC₅₀ = 0.3 μM against SNB-19 glioblastoma cells [15]
Mechanistic studies revealed that these compounds significantly reduced expression of CDKN1A in multiple tumor lines, with Compound 8 markedly downregulating BCL-2 expression in A549 and SNB-19 cells, indicating induction of apoptotic pathways [15].
Table 1: Experimental and Computational Lipophilicity Parameters of Selected Diquinothiazines
| Compound | Substituent | RM₀ (RP-TLC) | logPₜₗc | iLOGP | XLOGP3 | MLOGP | Cell Line (IC₅₀ μM) |
|---|---|---|---|---|---|---|---|
| 2 | Dimethylaminopropyl | 0.422 | 3.84 | 3.79 | 5.23 | 4.08 | A549 (0.3) |
| 8 | Pyrrolidinylethyl | 0.398 | 3.66 | 3.45 | 5.01 | 3.87 | SNB-19 (0.3) |
| 5 | Diethylaminoethyl | 0.411 | 3.78 | 3.52 | 5.34 | 4.12 | MDA-MB-231 (1.2) |
| 14 | Piperidinylpropyl | 0.435 | 3.94 | 4.01 | 5.67 | 4.33 | Caco-2 (2.1) |
Lipophilicity Determination Methodologies
Experimental RP-TLC Protocol
Stationary Phase:
- RP-18 silica gel plates (Merck, Germany)
Mobile Phase:
- Acetone–TRIS buffer (pH 7.4) mixtures with increasing buffer proportion
Experimental Procedure:
- Spot 0.1 μL of 1 mg/mL methanolic compound solutions on RP-18 plates
- Develop chromatograms in pre-saturated twin-trough chambers with mobile phase
- Dry plates and detect spots under UV light (254 nm and 365 nm)
- Measure retention factor (R𝐹) values for each compound
- Extrapolate to 0% organic modifier to obtain RM₀ values using the equation: R𝑀 = R𝑀⁰ + bC where C is the concentration of organic modifier in the mobile phase
- Convert RM₀ to logPₜₗc using reference compounds with known log P values [15]
Computational Log P Prediction
The researchers employed multiple algorithms for log P prediction:
- iLOGP: A physics-based method that uses free-energy perturbation
- XLOGP3: An atom-based additive method with correction factors
- MLOGP: A topological approach using molecular connectivity indices
- SILCOS-IT: A fragment-based method using group contributions [15]
Comparative analysis revealed that iLOGP demonstrated the closest correlation with experimental RM₀ values, highlighting its utility for early-stage drug candidate screening.
ADMET Property Predictions
Comprehensive ADMET profiling was conducted using SwissADME and pkCSM platforms, with key findings summarized below:
Table 2: Predicted ADMET Parameters for Lead Diquinothiazines
| Parameter | Compound 2 | Compound 8 | Optimal Range |
|---|---|---|---|
| GI Absorption | High | High | High |
| BBB Permeability | Yes | Yes | Target-dependent |
| CYP1A2 Inhibition | No | No | No inhibition |
| CYP2C9 Inhibition | Yes | No | No inhibition |
| CYP2D6 Inhibition | No | No | No inhibition |
| CYP3A4 Inhibition | Yes | Yes | No inhibition |
| Hepatotoxicity | No | No | No toxicity |
| AMES Toxicity | No | No | No mutagenicity |
| log S (ESOL) | -6.42 | -6.18 | > -6.0 |
| Skin Permeation | -6.82 cm/s | -6.95 cm/s | Low permeability |
The analysis confirmed that both lead compounds exhibited favorable ADMET profiles despite their relatively high lipophilicity, with good predicted gastrointestinal absorption and blood-brain barrier permeability – advantageous properties for targeting central nervous system cancers [15].
Experimental Workflow for Lipophilicity-Driven ADMET Optimization
The following diagram illustrates the comprehensive workflow for lipophilicity assessment and ADMET optimization employed in the diquinothiazine case study:
Diagram 1: Lipophilicity-ADMET Optimization Workflow (87 characters)
Structural Modification Strategies for Lipophilicity Optimization
The diquinothiazine case study demonstrates strategic approaches to fine-tune lipophilicity while maintaining target engagement:
Diagram 2: Lipophilicity Optimization Strategies (76 characters)
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Resources for Lipophilicity and ADMET Research
| Resource | Type | Primary Application | Key Features |
|---|---|---|---|
| RP-18 TLC Plates | Experimental | Chromatographic lipophilicity determination | High reproducibility, minimal compound requirement |
| SwissADME | Computational | In silico ADMET prediction | Free web tool, comprehensive descriptor calculation |
| pkCSM | Computational | Pharmacokinetic and toxicity prediction | Graph-based signatures, rapid screening capability |
| ADMET Predictor | Computational | Commercial ADMET platform | Proven correlation with experimental results [143] |
| PharmaBench | Data Resource | Benchmark dataset for ADMET models | 52,482 entries across 11 ADMET endpoints [20] |
| ChEMBL | Data Resource | Public bioactivity database | Manually curated SAR and property data [20] |
The optimization of diquinothiazine anticancer agents demonstrates the critical importance of systematic lipophilicity management in successful drug candidate design. Through the strategic application of complementary experimental and computational approaches, researchers achieved compounds with exceptional potency alongside favorable ADMET profiles.
The integration of chromatographic lipophilicity measurements with in silico ADMET prediction platforms provides a powerful framework for contemporary drug discovery, enabling researchers to identify potential liabilities early in the development process. As machine learning approaches continue to evolve, with AI-discovered drugs already showing improved clinical success rates [142], the precision of lipophilicity-ADMET relationships will further enhance our ability to design candidates with optimal pharmacokinetic and safety profiles.
This case study underscores that while lipophilicity remains a fundamental driver of compound behavior, its intelligent application within a holistic understanding of molecular properties provides the foundation for successful drug development in the modern era.
Evaluating the Performance of Multi-Task Learning Models for ADMET Prediction
The accurate prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties represents a critical challenge in modern drug discovery, with approximately 40-45% of clinical attrition still attributed to ADMET liabilities [47]. Within this context, lipophilicity serves as a fundamental molecular descriptor that significantly influences multiple ADMET endpoints, including solubility, permeability, metabolic stability, and volume of distribution [144] [79]. Traditional single-task learning (STL) approaches, which build separate models for each ADMET property, often struggle with data sparsity and fail to capture the complex interdependencies between properties influenced by shared underlying factors like lipophilicity [145] [146].
Multi-task learning (MTL) has emerged as a transformative paradigm that simultaneously predicts multiple ADMET endpoints by leveraging shared representations and commonalities across related prediction tasks [147] [145]. This technical guide comprehensively evaluates the performance of advanced MTL frameworks for ADMET prediction, with particular emphasis on how these models capture and utilize the influence of lipophilicity and other key molecular descriptors across pharmacokinetic and toxicological endpoints. By examining architectural innovations, task selection strategies, and representation learning techniques, this review provides drug development professionals with a rigorous assessment of MTL capabilities and limitations in predicting compound behavior in biological systems.
Foundations of Multi-Task Learning for ADMET Prediction
From Single-Task to Multi-Task Paradigms
Traditional ADMET prediction has predominantly relied on single-task learning models that follow a "one model, one task" approach [145]. While effective for endpoints with abundant labeled data, STL faces significant limitations when applied to the sparse, heterogeneous data landscape typical of ADMET assessment, where acquiring multiple experimental properties for the same compound is costly and time-consuming [147] [146]. The isolation of learning processes in STL fails to exploit the inherent connections between ADMET tasks, missing opportunities for knowledge transfer across related endpoints [145].
MTL addresses these limitations through a unified framework that solves multiple tasks simultaneously while exploiting commonalities and differences across ADMET endpoints [145]. During training, underlying knowledge among related ADMET endpoints can be transferred between them, compensating for scarce labels and improving generalization [147] [145]. This paradigm shift recognizes that ADMET properties are biologically interconnected—for instance, cytochrome P450 enzyme inhibition can increase plasma concentration (distribution), reduce clearance (excretion), and prolong half-life (excretion) of therapeutic agents [145].
The Critical Role of Lipophilicity in ADMET Properties
Lipophilicity, commonly measured as logP, serves as a master variable influencing multiple ADMET properties simultaneously [144] [79]. As a key component of Lipinski's Rule of 5, excessive lipophilicity (MlogP > 4.15) represents a primary risk factor for poor oral absorption [79]. Beyond absorption, lipophilicity strongly correlates with solubility, permeability, metabolic stability, plasma protein binding, and volume of distribution [79]. The ADMET Risk scoring system incorporates lipophilicity thresholds as part of its comprehensive assessment of compound developability, recognizing that lipophilic compounds often present higher risks for CYP metabolism and toxicity endpoints [79].
Recent research on imidazolone derivatives has demonstrated deliberate manipulation of lipophilic properties to optimize anticancer activity and ADMET profiles [144]. Compounds with dodecyl chains exhibited excellent efficacy against cancer cell lines, while maintaining favorable drug-likeness under Lipinski's guidelines [144]. These findings illustrate how strategic control of lipophilicity enables balancing potency with desirable ADMET characteristics—a balance that MTL models are particularly well-suited to predict through simultaneous consideration of multiple endpoints.
Advanced MTL Frameworks and Architectures
Key Architectural Innovations
Recent MTL frameworks for ADMET prediction have introduced substantial innovations in molecular representation, task balancing, and knowledge sharing. The table below summarizes the architectures and key features of prominent MTL models:
Table 1: Comparison of Advanced MTL Frameworks for ADMET Prediction
| Framework | Core Architecture | Molecular Representations | Task Weighting Approach | Key Innovations |
|---|---|---|---|---|
| MolP-PC [148] [149] | Multi-view fusion + MTL | 1D fingerprints, 2D molecular graphs, 3D geometric | Multi-task adaptive learning | Attention-gated fusion of multiple molecular views; achieved optimal performance in 27 of 54 tasks |
| QW-MTL [147] | Quantum-enhanced MTL | Quantum chemical descriptors + traditional features | Learnable exponential weighting | Incorporation of quantum chemical descriptors (dipole moment, HOMO-LUMO gap); outperformed STL in 12/13 TDC tasks |
| MTGL-ADMET [145] | "One primary, multiple auxiliaries" | Graph neural networks | Adaptive auxiliary task selection | Status theory + maximum flow for task selection; identifies crucial molecular substructures for each ADMET endpoint |
| Receptor.AI [150] | Multi-task deep learning | Mol2Vec embeddings + chemical descriptors | LLM-based consensus scoring | Combines graph-based embeddings with curated descriptors; predicts 38 human-specific ADMET endpoints |
Representation Learning Enhancements
The integration of multi-view molecular representations has significantly enhanced model capacity to capture structural and electronic determinants of ADMET properties. MolP-PC exemplifies this approach through parallel processing of 1D molecular fingerprints, 2D molecular graphs, and 3D geometric representations, with attention mechanisms selectively emphasizing the most relevant views for specific endpoints [148] [149]. This multi-view strategy addresses the limitation of single-molecule representations that often lead to information loss [148].
The QW-MTL framework incorporates quantum chemical descriptors including dipole moment, HOMO-LUMO gap, electron distributions, and total energy [147]. These physically-grounded 3D features capture molecular spatial conformation and electronic properties that are particularly relevant for ADMET outcomes influenced by lipophilicity, such as membrane permeability, metabolic susceptibility, and protein binding [147]. By enriching traditional 2D descriptors with quantum chemical features, QW-MTL provides a more comprehensive representation of the physicochemical determinants of ADMET properties.
Task Selection and Weighting Strategies
A critical advancement in MTL for ADMET prediction involves sophisticated approaches to task selection and loss balancing. The MTGL-ADMET framework introduces a novel "one primary, multiple auxiliaries" paradigm that strategically selects appropriate auxiliary tasks to enhance performance on a primary task of interest [145]. This approach utilizes status theory to identify friendly auxiliaries and maximum flow algorithms to estimate potential performance gains, effectively addressing the challenge of negative transfer where inappropriate task combinations degrade performance [145].
For loss balancing, QW-MTL implements an exponential task weighting scheme that combines dataset-scale priors with learnable parameters [147]. This dynamic adjustment of task contributions during training mitigates the imbalanced optimization problem common in MTL, where heterogeneous task difficulties and data scales can lead to dominant tasks suppressing weaker ones [147]. The learnable weighting mechanism enables the model to automatically prioritize tasks based on their learning status and importance, reducing the need for manual tuning of loss coefficients.
Experimental Protocols and Performance Benchmarking
Standardized Evaluation Frameworks
Rigorous benchmarking of MTL models requires standardized datasets, splitting strategies, and evaluation metrics. The Therapeutics Data Commons (TDC) has emerged as a widely adopted benchmark platform that provides curated ADMET datasets and standardized evaluation protocols [147]. Recent studies have increasingly adopted leaderboard-style data splits to ensure realistic assessment of model generalization, addressing concerns about inflated performance estimates from random splitting [147].
The MolP-PC framework underwent comprehensive evaluation across 54 ADMET tasks, with scaffold-based splitting ensuring that models are tested on structurally novel compounds not represented in training data [148] [149]. This approach provides a more realistic estimate of real-world performance where models must predict properties for novel chemical scaffolds beyond the training distribution.
Quantitative Performance Comparison
The table below summarizes the quantitative performance of advanced MTL frameworks compared to single-task baselines across key ADMET endpoints:
Table 2: Performance Comparison of MTL vs. Single-Task Models on ADMET Endpoints
| ADMET Endpoint | Metric | Single-Task Baseline | MTL Framework | Performance Gain | Lipophilicity Correlation |
|---|---|---|---|---|---|
| Human Intestinal Absorption (HIA) | AUC | 0.916 ± 0.054 [145] | 0.981 ± 0.011 [145] | +7.1% | High - critical for permeability |
| Oral Bioavailability (OB) | AUC | 0.716 ± 0.035 [145] | 0.749 ± 0.022 [145] | +4.6% | Moderate - multiple factors |
| P-gp Inhibition | AUC | 0.917 ± 0.006 [145] | 0.928 ± 0.008 [145] | +1.2% | High - lipophilic compounds favored |
| Platinum Complex Solubility | RMSE | 0.86 [151] | 0.62 [151] | -27.9% | Inverse correlation |
| Platinum Complex Lipophilicity | RMSE | N/A [151] | 0.44 [151] | N/A | Direct measurement |
| CYP450 Inhibition | AUC | 0.745 ± 0.029 [145] | 0.803 ± 0.024 [145] | +7.8% | High - metabolic stability |
MTL frameworks demonstrate particularly strong advantages for endpoints with limited training data, where knowledge transfer from related tasks substantially improves prediction accuracy [145] [149]. The MTGL-ADMET model achieved performance improvements across multiple endpoints, with the most significant gains observed for tasks benefiting from appropriate auxiliary task selection [145]. Additionally, MTL models show enhanced performance on novel chemical scaffolds, as evidenced by the improved prediction of platinum complex solubility when trained on extended chemical spaces [151].
Experimental Methodology for MTL Model Validation
Comprehensive validation of MTL models requires specialized experimental protocols that assess both individual endpoint accuracy and cross-task consistency:
Data Curation and Preprocessing: High-quality datasets with standardized assay protocols and normalized values are essential [47] [150]. The Apheris Federated ADMET Network implements rigorous data validation including sanity checks, assay consistency verification, and normalization procedures [47].
Task Selection and Grouping: MTGL-ADMET employs status theory and maximum flow algorithms to identify optimal auxiliary tasks for each primary prediction task [145]. This involves training individual and pairwise tasks to build a task association network before selecting auxiliary tasks that maximize potential performance gains.
Multi-View Feature Extraction: MolP-PC extracts 1D molecular fingerprints (ECFP6), 2D molecular graphs (atom and bond features), and 3D geometric representations (quantum chemical descriptors) for each compound [148] [149]. These multi-view features are integrated through attention-gated fusion mechanisms.
Multi-Task Adaptive Training: QW-MTL implements learnable exponential task weighting with dataset-scale priors to balance loss contributions across tasks [147]. Training typically employs cross-entropy loss for classification tasks and mean squared error for regression endpoints.
Generalization Assessment: Models are evaluated using rigorous scaffold-based splitting and time-split validation to assess performance on novel chemical classes [151]. Case studies on specific compound series (e.g., Oroxylin A, platinum complexes) demonstrate real-world applicability [148] [151].
Table 3: Key Research Reagents and Computational Tools for MTL in ADMET Prediction
| Resource Category | Specific Tools/Platforms | Function and Application | Relevance to Lipophilicity Research |
|---|---|---|---|
| Benchmark Datasets | TDC [147], MoleculeNet [145] | Standardized ADMET datasets for model training and benchmarking | Include curated logP measurements and related properties |
| Molecular Representation | RDKit [147], Mordred [150], Quantum Chemical Descriptors [147] | Generation of 1D/2D/3D molecular features and descriptors | Direct computation of logP, logD, and other lipophilicity metrics |
| Deep Learning Frameworks | Chemprop [147], D-MPNN [147], Graph Neural Networks [145] | Implementation of MTL architectures for ADMET endpoint prediction | Enable modeling of lipophilicity effects across multiple endpoints |
| Commercial Platforms | ADMET Predictor [79], Receptor.AI [150] | Enterprise-level ADMET prediction with GUI and API access | Incorporate lipophilicity in ADMET Risk scoring systems |
| Validation Tools | Scaffold Split Generators [47], Applicability Domain Assessment [79] | Assessment of model generalization and chemical space coverage | Evaluate performance across lipophilicity ranges |
| Federated Learning | Apheris [47], MELLODDY [47] | Collaborative training across distributed datasets | Expand chemical space coverage including diverse lipophilicity profiles |
Visualization of MTL Workflows and Lipophilicity Influences
Multi-task learning represents a paradigm shift in ADMET prediction that effectively addresses the interconnected nature of pharmacokinetic and toxicological properties. By leveraging shared representations and strategic task weighting, advanced MTL frameworks including MolP-PC, QW-MTL, and MTGL-ADMET have demonstrated superior performance compared to single-task approaches, particularly for endpoints with limited training data or strong dependencies on fundamental molecular properties like lipophilicity [148] [147] [145].
The integration of multi-view molecular representations encompassing 1D, 2D, and 3D features provides a more comprehensive characterization of the structural and electronic determinants of ADMET properties [148] [149]. Meanwhile, innovative task selection and weighting strategies mitigate the challenges of negative transfer and imbalanced optimization, enabling more effective knowledge sharing across related endpoints [147] [145]. These architectural advances, combined with rigorous benchmarking standards and federated learning approaches that expand chemical space coverage, position MTL as an indispensable tool for modern drug discovery [47].
For researchers and drug development professionals, the growing maturity of MTL frameworks offers powerful capabilities for predicting the complex interplay between lipophilicity and ADMET properties, enabling more efficient compound optimization and reduced late-stage attrition. As these models continue to evolve through incorporation of quantum chemical descriptors, interpretability enhancements, and expanded applicability domains, they promise to further accelerate the development of safer, more effective therapeutics.
Comparative ADMET Profiles of Compounds Across Different Lipophilicity Ranges
Within modern drug discovery, the optimization of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties is pivotal for mitigating late-stage attrition and ensuring the clinical success of new therapeutic agents [20] [152]. Among the various physicochemical descriptors, lipophilicity, most frequently quantified as the partition coefficient between n-octanol and water (LogP), stands as a principal parameter that profoundly influences a compound's pharmacokinetic behavior and safety profile [15]. This whitepaper, framed within a broader thesis on the impact of lipophilicity on ADMET properties research, provides a technical guide for scientists. It synthesizes current data, computational methodologies, and experimental protocols to elucidate the comparative ADMET profiles of compounds stratified by their lipophilicity, thereby offering a framework for more rational and efficient drug design.
The critical role of lipophilicity stems from its direct correlation with a molecule's ability to interact with biological membranes and proteins. Medicinal substances with moderate lipophilicity tend to demonstrate optimal absorption and distribution characteristics, while excessive lipophilicity often leads to poor aqueous solubility, heightened metabolic clearance, and increased risk of promiscuous binding and toxicity [15] [6]. Consequently, understanding the nuanced relationships between defined lipophilicity ranges and specific ADMET endpoints is essential for steering lead optimization campaigns toward chemical space with a higher probability of success.
The following tables summarize the definitive relationships between lipophilicity ranges and key ADMET parameters, drawing from consolidated experimental and in silico data.
Table 1: Impact of Lipophilicity on Fundamental Physicochemical and ADMET Properties
| Lipophilicity Range (LogP) | Solubility (LogS) | Absorption (Caco-2 Permeability, 10⁻⁶ cm/s) | Plasma Protein Binding (% Bound) | BBB Penetration Likelihood | Primary Metabolic Route |
|---|---|---|---|---|---|
| Low (LogP < 1) | High (> -4) | Low (< 10) | Low (< 70%) | Unlikely | Renal Clearance / Phase II Conjugation |
| Moderate (LogP 1-3) | Moderate (-4 to -6) | Moderate to High (50 - 150) | Moderate (70-90%) | Probable | Mixed (CYP & UGT) |
| High (LogP 3-5) | Low (-6 to -8) | High (> 150) | High (> 90%) | Highly Likely | CYP-Mediated Oxidation (e.g., CYP3A4) |
| Very High (LogP > 5) | Very Low (< -8) | Variable (may be limited by solubility) | Very High (> 95%) | Highly Likely | High Metabolic Lability / Poor Clearance |
Table 2: Toxicity and Clinical Risk Association with Increasing Lipophilicity
| Lipophilicity Range (LogP) | hERG Inhibition Risk | Hepatotoxicity Risk | Clinical Attrition Risk | Key Risks and Challenges |
|---|---|---|---|---|
| Low (LogP < 1) | Low | Low | Low (but often lacks efficacy) | Poor membrane permeability, limited tissue distribution, rapid renal clearance. |
| Moderate (LogP 1-3) | Low to Moderate | Low to Moderate | Optimal Range | Balanced properties; generally desirable for oral drugs. |
| High (LogP 3-5) | Moderate to High | Elevated | High | Low solubility, metabolic instability, promiscuous target interactions. |
| Very High (LogP > 5) | High | High | Very High | Extreme insolubility, high metabolic clearance, accumulation in tissues, significant toxicity risk. |
The data in these tables corroborate findings from case studies, such as that of ACP-105, a high-logP compound (LogP 3.0–3.52) which demonstrated low solubility (LogS ~ -4.1 to -4.4), high plasma protein binding (77–99%), and a metabolic profile dominated by CYP3A4-mediated oxidation [153]. This profile is characteristic of the challenges faced in the high lipophilicity range.
Experimental and In Silico Methodologies for Profiling
A multi-faceted approach, combining robust experimental measurements with advanced in silico predictions, is required to accurately characterize the ADMET profiles of compounds across the lipophilicity spectrum.
Determining Lipophilicity: Experimental Protocols
3.1.1 Reversed-Phase Thin-Layer Chromatography (RP-TLC) This chromatographic technique is a widely used indirect method for determining lipophilicity parameters (RM0) [15].
- Stationary Phase: RP-18 silica plates.
- Mobile Phase: Acetone–TRIS buffer (pH 7.4) at varying ratios to establish a relationship between mobile phase composition and RM value [15].
- Procedure:
- Spot the compound solution on the TLC plate.
- Develop the plate in a saturated chromatographic chamber with the mobile phase.
- Dry the plate and detect the compound spots (e.g., under UV light).
- Calculate the RM value. The extrapolated RM0 value (for 0% organic modifier) serves as the chromatographic descriptor of lipophilicity.
- Advantages: Requires a small amount of sample, is cost-effective, and allows for high-throughput analysis.
3.1.2 Shake-Flask Method This is the classic benchmark technique for the direct measurement of the partition coefficient [15].
- Procedure:
- Dissolve the purified compound in a mixture of n-octanol and a buffer (e.g., phosphate buffer, pH 7.4), pre-saturated with each other.
- Shake the mixture vigorously for a defined period (1-24 hours) to reach partitioning equilibrium.
- Separate the two phases by centrifugation.
- Quantify the concentration of the compound in each phase using a suitable analytical method (e.g., HPLC-UV).
- Calculate LogP as the logarithm of the ratio of concentrations in n-octanol to the buffer.
- Advantages: Considered a gold standard.
- Disadvantages: Time-consuming, requires relatively large amounts of pure compound, and is limited to a LogP range of approximately -2 to 4 [15].
Predicting ADMET Properties: A Guide to In Silico Tools
Machine learning (ML) and artificial intelligence (AI) have revolutionized the early prediction of ADMET properties [152] [81]. The following workflow diagram illustrates the integration of these methodologies in a drug discovery pipeline.
Diagram 1: An AI/ML workflow for ADMET prediction in drug discovery.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Software and Platforms for In Silico ADMET Profiling
| Tool Name | Type/Function | Key Features & Applications |
|---|---|---|
| SwissADME | Web Tool | Calculates physicochemical descriptors, predicts pharmacokinetics, and evaluates drug-likeness; useful for rapid, initial profiling [153] [15]. |
| pkCSM | Web Tool | Predicts a wide range of ADMET-related endpoints, including permeability, metabolism, and toxicity parameters [153] [15]. |
| ADMETlab 3.0 | Web Platform | Comprehensive platform predicting over 100 ADMET endpoints; integrates multi-task learning for improved accuracy [153] [150]. |
| Chemaxon | Software Suite | Provides high-performance predictive models for key properties like LogP, pKa, and solubility; includes chemicalize toolkit [6]. |
| ADMET Predictor | Commercial Software | Machine learning tool for predicting >175 ADMET endpoints; includes metabolic hotspot identification [153]. |
| RDKit | Cheminformatics Library | Open-source toolkit for calculating molecular descriptors and fingerprints; serves as the backend for many ML models [101]. |
| PharmaBench | Benchmark Dataset | Open-source dataset for developing AI models; comprises eleven ADMET datasets with over 52,000 entries [20]. |
The convergence of these computational tools with experimental validation creates a powerful feedback loop. As illustrated in Diagram 1, predictions guide experimental priorities, and experimental results, in turn, refine and validate the computational models, leading to more informed lead optimization cycles [81] [154].
Integrated Analysis and Future Directions
The interplay between lipophilicity and ADMET properties is complex and nonlinear. While the tables in Section 2 provide a generalized guide, the precise impact of LogP is modulated by other molecular features such as molecular weight, topological polar surface area (TPSA), and the number of hydrogen bond donors and acceptors [152] [6]. For instance, a high-logP compound might still demonstrate acceptable solubility if it has a high TPSA or exists as an amorphous solid.
The future of ADMET profiling lies in the deeper integration of multimodal data and the adoption of more sophisticated AI-driven models. Next-generation approaches are moving beyond simple QSAR models to leverage graph neural networks (GNNs), ensemble learning, and multitask frameworks that can capture the complex, high-dimensional relationships between structure, properties, and biological activity [152] [154] [150]. Furthermore, the push for model interpretability—moving beyond "black-box" predictions—is crucial for gaining scientific insight and regulatory acceptance [150]. Initiatives like the FDA's plan to phase out animal testing for certain cases in favor of New Approach Methodologies (NAMs), which include validated AI models, underscore the growing importance of robust in silico profiling [150].
In conclusion, a disciplined approach to managing lipophilicity remains a cornerstone of successful drug design. By systematically leveraging the experimental and computational toolkit detailed in this guide, researchers can more effectively navigate the ADMET landscape, de-risk candidates earlier in the development process, and accelerate the delivery of safer and more efficacious therapeutics.
Within the paradigm of modern drug discovery, the lipophilicity of a candidate molecule stands as a pivotal physicochemical property that profoundly influences its Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET). The logarithm of the partition coefficient (LogP) or distribution coefficient (LogD) at physiological pH 7.4 (Log D7.4) serves as the standard quantitative descriptor for lipophilicity, measuring the compound's affinity for lipophilic versus aqueous environments [83]. A compelling body of evidence links this single parameter to critical pharmacokinetic behaviors and clinical outcomes, positioning it as a key determinant in the transition from preclinical findings to human results. This technical guide examines the established correlations between preclinical lipophilicity measurements and their tangible impacts on human pharmacokinetics and safety, providing a framework for researchers to leverage this relationship in optimizing drug candidates.
Quantitative Correlations: From Preclinical Data to Clinical Outcomes
Lipophilicity and Drug-Induced Liver Injury (DILI) Risk
Analysis of large-scale datasets from the FDA-approved drug portfolio has revealed statistically significant associations between lipophilicity, daily dose, and the risk of Drug-Induced Liver Injury (DILI). The "Rule of Two" (RO2) model identifies compounds with high lipophilicity (LogP ≥ 3) administered at high daily doses (≥ 100 mg) as being at substantially increased DILI risk [155].
Table 1: Association between Lipophilicity, Daily Dose, and DILI Risk Across Independent Annotations
| Annotation Dataset | DD ≥100 mg & LogP ≥3 (RO2) Odds Ratio | Statistical Significance (p-value) |
|---|---|---|
| Chen et al. | 5.79 | p < 0.05 |
| Greene et al. | 11.50 | p < 0.05 |
| Xu et al. | 2.32 | p < 0.05 |
| Sakatis et al. | 4.21 | p < 0.05 |
| Zhu et al. | 6.18 | p < 0.05 |
This combination of high lipophilicity and high daily dose consistently demonstrated a significant association with DILI risk across all five independent annotation datasets, with odds ratios ranging from 2.32 to 11.50 [155]. The relationship between lipophilicity alone and DILI risk was weaker and inconsistent across datasets, underscoring the importance of considering dose as a co-factor in risk assessment.
Lipophilicity-Driven Clearance Pathways and Toxicity Profiles
Preclinical studies demonstrate that strategic modification of a compound's lipophilicity can directly modulate its in vivo biodistribution and clearance route, thereby improving its therapeutic index. Research on targeted alpha-particle therapy (TAT) conjugates for metastatic melanoma established a clear correlation between lipophilicity (Log D7.4) and kidney uptake [83].
Table 2: Impact of Lipophilicity on Biodistribution and Toxicity of a Peptide-Drug Conjugate
| Linker Lipophilicity (Log D7.4) | Kidney Uptake | Kidney Toxicity | Primary Clearance Route |
|---|---|---|---|
| Lower Lipophilicity | Increased | Acute nephropathy and death | Renal |
| Higher Lipophilicity | Decreased | Chronic progressive nephropathy | Hepatic |
Tuning the lipophilicity of the radiopharmaceutical through linker modification effectively shifted the primary clearance from renal to hepatic, simultaneously decreasing kidney uptake, absorbed radiation dose, and associated acute toxicity [83]. This demonstrates a direct preclinical-to-clinical translation where a physicochemical property directly informed the design of a safer therapeutic.
Experimental Protocols for Lipophilicity and ADMET Assessment
Determination of Distribution Coefficient (Log D)
The distribution coefficient (Log D) is a critical measurement that reflects the lipophilicity of a compound at a specified pH, typically physiological pH 7.4.
Protocol: Shake-Flask Method for Log D7.4 [156]
- Solution Preparation: Pre-saturate phosphate-buffered saline (PBS, pH 7.4) and n-octanol by mixing them in equal volumes (e.g., 2 mL each) and agitating them overnight on a vibrating shaker (e.g., 2500 rpm) at 25°C to achieve equilibrium.
- Compound Introduction: Spike the DMSO stock solution of the test compound directly into the organic (n-octanol) layer of the equilibrated mixture.
- Incubation and Partitioning: Incubate the mixture at 25°C for 4 hours with continuous shaking to allow the compound to partition between the two immiscible phases.
- Sample Collection: After incubation and phase separation, withdraw precise aliquots (e.g., 100 µL) from both the organic and aqueous layers.
- Quantitative Analysis: Dilute samples with an appropriate solvent (e.g., 80:20 methanol:water) containing an internal standard. Analyze the concentration of the compound in each phase using a sensitive quantitative method such as LC-MS/MS.
- Calculation: Calculate the Log D7.4 value using the following equation:
- Log D7.4 = log₁₀ ( [Compound]ₒᵣ𝑔ₐₙᵢ𝒸 / [Compound]ₐ𝑞ᵤₑₒᵤₛ )
where
[Compound]ₒᵣ𝑔ₐₙᵢ𝒸is the concentration in the n-octanol layer and[Compound]ₐ𝑞ᵤₑₒᵤₛis the concentration in the PBS layer [156].
- Log D7.4 = log₁₀ ( [Compound]ₒᵣ𝑔ₐₙᵢ𝒸 / [Compound]ₐ𝑞ᵤₑₒᵤₛ )
where
In Vitro Assays for Key ADMET Endpoints
Standardized in vitro assays form the backbone of preclinical ADMET profiling. Key assays relevant to lipophilicity-driven outcomes include:
- Liver Microsomal Stability [157]
- Objective: To measure the metabolic stability of a compound by assessing its degradation rate in mouse (MLM) or human (HLM) liver microsomes.
- Application: Serves as an in vitro estimate of in vivo metabolic clearance and intrinsic half-life.
- Cell-Based Permeability Assay (MDR1-MDCKII) [157]
- Objective: To model cell layer permeation, including critical barriers like the blood-brain barrier (BBB), using Madin-Darby Canine Kidney cells transfected with the human MDR1 gene.
- Application: Predicts a compound's ability to permeate cellular barriers, a property highly influenced by lipophilicity.
- Plasma Protein Binding (PPB) [156]
- Objective: To determine the fraction of a drug bound to plasma proteins versus the free, unbound fraction, using methods like Rapid Equilibrium Dialysis (RED).
- Application: Critical for understanding distribution and efficacy, as only the free fraction is considered pharmacologically active.
Lipophilicity in ADMET Pathway
Table 3: Key Research Reagents and Platforms for ADMET and Lipophilicity Studies
| Reagent / Platform | Function / Application | Relevance to Lipophilicity-ADMET |
|---|---|---|
| Caco-2 Cells | In vitro model of human intestinal permeability [156] | Predicts absorption, highly influenced by compound lipophilicity. |
| MDR1-MDCKII Cells | Cell line modeling blood-brain barrier and cellular permeation [157] | Assesses brain penetration and P-gp efflux, key for CNS-targeted drugs. |
| Human/Rat Liver Microsomes | In vitro metabolic stability assessment (HLM/MLM) [157] [156] | Estimates metabolic clearance rate; lipophilicity affects enzyme access. |
| Rapid Equilibrium Dialysis (RED) Device | Determines plasma protein binding fraction [156] | Quantifies free drug concentration; lipophilicity influences binding affinity. |
| ADMET Prediction Platforms (e.g., ADMETlab, SwissADME) | In silico prediction of ADMET properties [45] [150] | Uses algorithms (e.g., Mol2Vec) to model properties from structure, including LogP. |
Integrated Workflow for Preclinical Profiling
Integrated ADMET Profiling Workflow
The correlation between preclinical lipophilicity and human outcomes is a powerful tool in the drug developer's arsenal. By integrating robust experimental Log D determination with a suite of targeted in vitro and in vivo ADMET assays, researchers can build predictive models of human pharmacokinetics and toxicity. Adherence to frameworks like the "Rule of Two," which integrates lipophilicity with dose, provides a validated strategy for de-risking candidates. As artificial intelligence and machine learning models continue to advance, leveraging larger and more standardized datasets, the precision of these predictions will only increase [150] [101] [154]. Ultimately, a disciplined, data-driven approach to understanding and applying lipophilicity relationships is fundamental to designing safer, more effective drugs and improving clinical success rates.
Conclusion
Lipophilicity remains an indispensable, master variable in the rational design of drugs with desirable ADMET properties. A deep understanding of its foundational impact, coupled with robust methodological tools for its assessment, allows researchers to proactively troubleshoot issues and optimize candidate compounds. The future of lipophilicity optimization lies in the sophisticated integration of advanced in silico models, including graph-based machine learning and multi-task learning, with high-quality experimental data. As drug targets become more challenging, such as in protein-protein interactions and CNS disorders, the strategic management of lipophilicity will be paramount. Embracing a holistic, multi-parameter optimization framework that carefully balances lipophilicity with other key properties is the definitive path to reducing attrition rates and accelerating the development of safer, more effective therapeutics.
References
- 1. The impact of lipophilicity on environmental processes ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X18315340]
- 2. LogP vs LogD - What is the Difference? [https://www.acdlabs.com/blog/partitioning-logp-or-logd-are-you-using-measuring-the-right-descriptor/]
- 3. Introduction to Pharmacokinetics: Four Steps in a Drug's ... [https://genomind.com/providers/introduction-to-pharmacokinetics-four-steps-in-a-drugs-journey-through-the-body/]
- 4. LogP, LogD, pKa and LogS: A Physicists guide to basic ... [https://doktormike.gitlab.io/posts/navigating-logp-logd-pka-and-logs-a-physicists-guide/]
- 5. Study of Lipophilicity and ADME Properties of 1,9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10138389/]
- 6. Key Properties in Drug Design | Predicting Lipophilicity, ... [https://chemaxon.com/blog/presentation/key-properties-in-drug-design-predicting-lipophilicity-pka-and-solubility-0]
- 7. Lipophilicity - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/lipophilicity]
- 8. Robust assessment of statistical significance in the use ... [https://pubmed.ncbi.nlm.nih.gov/10640503/]
- 9. Lipophilicity, Pharmacokinetic Properties, and Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8224687/]
- 10. Impact of Fluorine Pattern on Lipophilicity and Acid–Base ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12124721/]
- 11. Study of the Lipophilicity and ADMET Parameters of New Anticancer Diquinothiazines with Pharmacophore Substituents - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11206290/]
- 12. Lipophilicity and its relationship with passive drug ... [https://pubmed.ncbi.nlm.nih.gov/21052797/]
- 13. Utility of Physicochemical Properties for the Prediction of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7025387/]
- 14. Liquid chromatography in determination of ... [https://www.sciencedirect.com/science/article/abs/pii/S1570023225001266]
- 15. Study of the Lipophilicity and ADMET Parameters of New ... [https://www.mdpi.com/1424-8247/17/6/725]
- 16. Lipophilic Studies and In Silico ADME Profiling of ... [https://www.mdpi.com/1422-0067/24/15/12230]
- 17. Enhancing Drug Discovery: Autoencoder-Based Latent ... [https://arxiv.org/html/2506.00223v1]
- 18. Use of TLC and Computational Methods to Determine ... [https://pubmed.ncbi.nlm.nih.gov/41011127/]
- 19. Evaluation of ADMET Predictor in Early Discovery Drug ... [https://pubmed.ncbi.nlm.nih.gov/34750195/]
- 20. PharmaBench: Enhancing ADMET benchmarks with large ... [https://www.nature.com/articles/s41597-024-03793-0]
- 21. From Patterns to Pills: How Informatics Is Shaping Medicinal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114992/]
- 22. High lipophilicity decreases drug transport across intestinal ... [https://pubmed.ncbi.nlm.nih.gov/8182532/]
- 23. Quantitative analysis for lipophilic drug transport through a ... [https://www.sciencedirect.com/science/article/abs/pii/S0928098719301642]
- 24. An industrial procedure for the intestinal permeability ... [https://www.nature.com/articles/s41598-023-47306-2]
- 25. Lipophilicity - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/chemistry/lipophilicity]
- 26. Drug Distribution - StatPearls - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK567736/]
- 27. Effect of Drug Lipophilicity and Ionization on Permeability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2976956/]
- 28. Plasma Protein Binding - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/plasma-protein-binding]
- 29. Plasma protein binding [https://en.wikipedia.org/wiki/Plasma_protein_binding]
- 30. 3.3 Drug distribution and plasma protein binding [https://fiveable.me/introduction-to-pharmacology/unit-3/drug-distribution-plasma-protein-binding/study-guide/qdxVz0bIhPJExOCj]
- 31. Mechanisms of penetration and diffusion of drugs ... [https://www.sciencedirect.com/science/article/pii/S0939641124002200]
- 32. Volume of distribution [https://derangedphysiology.com/main/cicm-primary-exam/pharmacokinetics/Chapter-202/volume-distribution]
- 33. Precision targeting of the CNS: recent progress in brain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278267/]
- 34. The blood–brain barrier: Structure, regulation and drug ... [https://www.nature.com/articles/s41392-023-01481-w]
- 35. THE ROLE OF BBB EFFLUX PUMPS IN DRUG ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7715363/]
- 36. The influence of lipophilicity in drug discovery and design [https://pubmed.ncbi.nlm.nih.gov/22992175/]
- 37. Lipophilicity - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/lipophilicity]
- 38. Drug efflux transporters in the CNS [https://www.sciencedirect.com/science/article/abs/pii/S0169409X02001722]
- 39. Advances in brain-targeted delivery strategies and natural ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03415-w]
- 40. A new era in brain drug delivery: Integrating multivalency ... [https://www.sciencedirect.com/science/article/pii/S0169409X2500122X]
- 41. Drug interactions due to cytochrome P450 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1312247/]
- 42. The Effect of Cytochrome P450 Metabolism on Drug ... [https://www.aafp.org/pubs/afp/issues/2007/0801/p391.html]
- 43. metabolic stability, CYP inhibition and induction [https://www.sciencedirect.com/science/article/abs/pii/S1740674904000459]
- 44. FDA's Examples of Drugs that Interact with CYP Enzymes ... [https://www.fda.gov/drugs/drug-interactions-labeling/healthcare-professionals-fdas-examples-drugs-interact-cyp-enzymes-and-transporter-systems]
- 45. Virtual screening, ADMET prediction, molecular docking ... [https://www.nature.com/articles/s41598-024-75292-6]
- 46. In Silico drug evaluation by molecular docking, ADME ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12135494/]
- 47. Federated Learning for ADMET Prediction [https://www.apheris.com/resources/blog/federated-learning-for-admet-prediction-expanding-model-applicability]
- 48. Advancing ADMET prediction through multiscale fragment- ... [https://pubmed.ncbi.nlm.nih.gov/41021261/]
- 49. Lipophilicity and ADMET Analysis of Quinoline-1,4-quinone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9867208/]
- 50. Computational analysis of calculated physicochemical and ... [https://www.nature.com/articles/srep46277]
- 51. hERG toxicity prediction in early drug discovery using ... [https://www.nature.com/articles/s41598-025-99766-3]
- 52. hERG toxicity assessment: Useful guidelines for drug design [https://pubmed.ncbi.nlm.nih.gov/32283295/]
- 53. Critical heart failure associated with beta-blocker-induced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11630001/]
- 54. Drug-induced phospholipidosis [https://www.medsafe.govt.nz/profs/PUArticles/March2024/Drug-induced-phospholipidosis.html]
- 55. Drug-induced phospholipidosis caused by combinations of ... [https://www.sciencedirect.com/science/article/abs/pii/S0887233316300947]
- 56. A machine learning and live-cell imaging tool kit uncovers ... [https://www.sciencedirect.com/science/article/pii/S2451945623003227]
- 57. Liquid Chromatography on the Different Methods for ... [https://www.mdpi.com/2227-9040/10/8/340]
- 58. Comparison of the Utility of RP-TLC Technique and Different ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6749294/]
- 59. Setup and validation of shake-flask procedures for the ... [https://www.sciencedirect.com/science/article/abs/pii/S0928098715002043]
- 60. Application of reversed-phase thin layer chromatography ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708519308076]
- 61. Reversed Phase Thin Layer Chromatography - an overview [https://www.sciencedirect.com/topics/chemistry/reversed-phase-thin-layer-chromatography]
- 62. Thin Layer Chromatography [https://chem.libretexts.org/Ancillary_Materials/Demos_Techniques_and_Experiments/General_Lab_Techniques/Thin_Layer_Chromatography]
- 63. Lipophilicity of Drug [https://www.bocsci.com/blog/lipophilicity-of-drug/?srsltid=AfmBOorBPSEjR2s1l8skZ5RLDRxPjF_pelUMe7ALnynd104fu6mt_Xzd]
- 64. Effect of lipophilicity on drug distribution and elimination [https://pubmed.ncbi.nlm.nih.gov/33450083/]
- 65. Computational Approaches in Preclinical Studies on Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7517894/]
- 66. SwissADME: a free web tool to evaluate pharmacokinetics ... [https://www.nature.com/articles/srep42717]
- 67. FAQ [https://www.swissadme.ch/faq.php]
- 68. pkCSM - pharmacokinetics [https://biosig.lab.uq.edu.au/pkcsm/]
- 69. ADME QSAR Models - StarDrop [https://optibrium.com/products/stardrop/modules/adme-qsar/]
- 70. StarDrop Small Molecule Drug Discovery & Data ... [https://optibrium.com/products/stardrop/]
- 71. ADMET property prediction: The state of the art and current ... [https://optibrium.com/knowledge-base/admet-property-prediction-the-state-of-the-art-and-current-challenges/]
- 72. Study of Lipophilicity and ADME Properties of 1,9- ... [https://pubmed.ncbi.nlm.nih.gov/37108135/]
- 73. Analysis of Lipophilicity and Pharmacokinetic Parameters of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11435374/]
- 74. Lipophilicity of Drug [https://www.bocsci.com/blog/lipophilicity-of-drug/?srsltid=AfmBOorUfxCTnKFHal7LqAlhvnJwnBdR-cnAmP2jtRII-ZSaRRMKKpuQ]
- 75. 10H-1,9-diazaphenothiazine and its 10-derivatives [https://pmc.ncbi.nlm.nih.gov/articles/PMC6691808/]
- 76. Methods for Determination of Lipophilicity [https://encyclopedia.pub/entry/26444]
- 77. Evaluation of the Lipophilicity of Angularly Condensed Diquino [https://pmc.ncbi.nlm.nih.gov/articles/PMC11013424/]
- 78. Lipophilicity and ADMET Analysis of Quinoline-1,4- ... [https://www.mdpi.com/1999-4923/15/1/34]
- 79. ADMET Predictor® Modules [https://www.simulations-plus.com/software/admetpredictor/]
- 80. Quantum chemical modeling, molecular docking, and ... [https://www.nature.com/articles/s41598-025-90495-1]
- 81. Leveraging machine learning models in evaluating ADMET ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12205928/]
- 82. Molecular inflation, attrition and the rule of five [https://www.sciencedirect.com/science/article/abs/pii/S0169409X16300370]
- 83. Lipophilicity Determines Routes of Uptake and Clearance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8033749/]
- 84. A drug-likeness toolbox facilitates ADMET study in ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644619304180]
- 85. Lipinski's rule of five [https://en.wikipedia.org/wiki/Lipinski%27s_rule_of_five]
- 86. Lipinski's Rule of Five - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/lipinskis-rule-of-five]
- 87. Drug-like Properties and Fraction Lipophilicity Index as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8920096/]
- 88. druglikeFilter 1.0: An AI powered filter for collectively ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12268052/]
- 89. A Method for Measuring the Lipophilicity of Compounds in ... [https://www.sciencedirect.com/science/article/pii/S2472555222077619]
- 90. Predicting ADMET Properties from Molecule SMILE: A Bottom-Up Approach Using Attention-Based Graph Neural Networks - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11207804/]
- 91. Advancing ADMET prediction for major CYP450 isoforms [https://biomedical-engineering-online.biomedcentral.com/articles/10.1186/s12938-025-01412-6]
- 92. Improved GNNs for Log D7.4 Prediction by Transferring Knowledge from Low-Fidelity Data - PubMed [https://pubmed.ncbi.nlm.nih.gov/37000044/]
- 93. Improved Lipophilicity and Aqueous Solubility Prediction with Composite Graph Neural Networks - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8539502/]
- 94. ADMET property prediction via multi-task graph learning ... [https://www.sciencedirect.com/science/article/pii/S2589004223023623]
- 95. Improving ADME Prediction with Multitask Graph Neural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12606635/]
- 96. AI trends in 2025: Graph Neural Networks [https://assemblyai.com/blog/ai-trends-graph-neural-networks]
- 97. (PDF) Lipophilicity Indices for Drug Development [https://www.academia.edu/31743711/Lipophilicity_Indices_for_Drug_Development]
- 98. Novel Strategies for the Formulation of Poorly Water-Soluble ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388907/]
- 99. Solubilization techniques used for poorly water-soluble drugs [https://www.sciencedirect.com/science/article/pii/S2211383524003484]
- 100. ADMET-score – a comprehensive scoring function for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6350845/]
- 101. Benchmarking ML in ADMET predictions: the practical impact ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01041-0]
- 102. Innovative applications and future perspectives of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11979114/]
- 103. Why Decreasing Lipophilicity Alone Is Often Not a Reliable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6004569/]
- 104. Probing the links between in vitro potency, ADMET and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6317702/]
- 105. admetSAR3.0: a comprehensive platform for exploration ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11223829/]
- 106. Predicting ADMET Properties from Molecule SMILE [https://www.mdpi.com/1999-4923/16/6/776]
- 107. Strategies for Enhancing the Permeation of CNS-Active Drugs through the Blood-Brain Barrier: A Review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6100436/]
- 108. Evolving Drug Delivery Strategies to Overcome the Blood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4900538/]
- 109. The penetration of therapeutics across the blood-brain barrier [https://pmc.ncbi.nlm.nih.gov/articles/PMC11604479/]
- 110. Progress in Brain Penetration Evaluation in Drug Discovery ... [https://www.sciencedirect.com/science/article/abs/pii/S0022356524343952]
- 111. Overcoming Challenges in Small-Molecule Drug Bioavailability [https://pmc.ncbi.nlm.nih.gov/articles/PMC11642056/]
- 112. Establishment and Application of Reversed-Phase Liquid ... [https://dmpkservice.wuxiapptec.com/articles/36-rapid-determination-of-lipophilicity-establishment-and-application-of-reversed-phase-liquid-chromatography-rp-hplc/]
- 113. Breaking Barriers: Medicinal Chemistry Strategies and ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425009845]
- 114. Experimental and computational analysis of lipophilicity ... [https://www.sciencedirect.com/science/article/abs/pii/S1570023225000339]
- 115. Molecular Modification: A Strategy in Drug Discovery and ... [https://biomedres.us/fulltexts/BJSTR.MS.ID.008220.php]
- 116. Prodrugs and their activation mechanisms for brain drug delivery [https://pubs.rsc.org/en/content/articlehtml/2025/md/d4md00788c]
- 117. Prodrugs for Improved Drug Delivery: Lessons Learned ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7692606/]
- 118. Prodrug Strategies in Medicinal Chemistry [https://www.acs.org/acs-webinars/library/prodrugs2.html]
- 119. Structure-guided dual modification prodrug strategy for ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517325007458]
- 120. Holistic drug design for multiparameter optimization in ... [https://www.sciencedirect.com/science/article/abs/pii/S0960894X21002298]
- 121. Multi-parameter optimization: identifying high quality ... [https://pubmed.ncbi.nlm.nih.gov/22316157/]
- 122. Which ADMET properties are important for me to predict? [https://optibrium.com/knowledge-base/blog-which-admet-properties-are-important-for-me-to-predict/]
- 123. Review article An overview of compound properties ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523423002660]
- 124. STELLA provides a drug design framework enabling ... [https://www.nature.com/articles/s41598-025-12685-1]
- 125. An In Silico Explainable Multiparameter Optimization ... [https://pubmed.ncbi.nlm.nih.gov/35581002/]
- 126. Challenges in Discovering Drugs That Target the Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8835748/]
- 127. New insights into protein–protein interaction modulators in ... [https://www.nature.com/articles/s41392-024-02036-3]
- 128. Study of the Lipophilicity of Tetracyclic Anticancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384454/]
- 129. Structure-based evolution of a promiscuous inhibitor to a ... [https://www.nature.com/articles/s41467-020-17741-0]
- 130. Quantum Mechanics-Driven Structure-Activity Relationship ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425007445]
- 131. Quantum mechanics-driven structure-activity relationship ... [https://pubmed.ncbi.nlm.nih.gov/40749258/]
- 132. Recent progress and future challenges in structure-based ... [https://www.sciencedirect.com/science/article/pii/S1525001625002771]
- 133. Comprehensive benchmarking of computational tools for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00931-z]
- 134. ADMET Benchmark Group [https://www.emergentmind.com/topics/admet-benchmark-group]
- 135. Lipophilicity prediction of peptides and peptide derivatives by consensus machine learning - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6151477/]
- 136. Use of TLC and Computational Methods to Determine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12472637/]
- 137. Understanding Lipinski's Rule of 5 and the Role of LogP ... [https://www.sailife.com/understanding-lipinskis-rule-of-5-and-the-role-of-logp-value-in-drug-design-and-development/]
- 138. Calculation of Molecular Lipophilicity: State-of-the-Art and ... [https://www.sciencedirect.com/science/article/pii/S0022354916329082]
- 139. Dimensionally reduced machine learning model for predicting ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-022-00660-1]
- 140. Study Models of Drug–Drug Interactions Involving P ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10673607/]
- 141. Prediction of drug-drug interaction risk of P-glycoprotein ... [https://www.sciencedirect.com/science/article/abs/pii/S1347436724000144]
- 142. How successful are AI-discovered drugs in clinical trials? A ... [https://www.sciencedirect.com/science/article/pii/S135964462400134X]
- 143. Evaluation of ADMET Predictor in Early Discovery Drug ... [https://www.semanticscholar.org/paper/Evaluation-of-ADMET-Predictor-in-Early-Discovery-Sohlenius-Sternbeck-Terelius/ebd10e73be2bca356214b9426b0af4d38c4ad8ee]
- 144. Design, synthesis, anticancer properties, and molecular ... [https://pubmed.ncbi.nlm.nih.gov/40425784/]
- 145. ADMET property prediction via multi-task graph learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10654589/]
- 146. Leveraging machine learning models in evaluating ADMET ... [https://pubmed.ncbi.nlm.nih.gov/40585410/]
- 147. Quantum-Enhanced Multi - Task with Learnable Weighting for... Learning [https://arxiv.org/html/2509.04601v1]
- 148. MolP-PC: a multi-view fusion and multi-task learning ... [https://www.sciencedirect.com/science/article/abs/pii/S1875536425609459]
- 149. MolP-PC: a multi-view fusion and multi - task framework for... learning [https://zgtryw.xml-journal.net/article/doi/10.1016/S1875-5364(25)60945-9]
- 150. Advanced ADMET Modeling Using Deep Learning and AI [https://www.technologynetworks.com/drug-discovery/blog/beyond-black-box-models-advances-in-data-driven-admet-modeling-403114]
- 151. Online OCHEM multi-task model for solubility and ... [https://www.sciencedirect.com/science/article/pii/S0162013425000704]
- 152. Machine learning approaches for next-generation ADMET ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625002004]
- 153. First multifaceted ADME profile of ACP-105 (CAS [https://pmc.ncbi.nlm.nih.gov/articles/PMC12534238/]
- 154. Revolutionizing pharmacology: AI-powered approaches in ... [https://www.sciencedirect.com/science/article/pii/S259009862500020X]
- 155. Associations of Drug Lipophilicity and Extent of Metabolism ... [https://www.mdpi.com/1422-0067/18/7/1335]
- 156. Preclinical pharmacokinetics and in vitro ADME properties ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12103360/]
- 157. antiviral-admet-2025 [https://polarishub.io/competitions/asap-discovery/antiviral-admet-2025]