Decoding Protein-Ligand Interactions: How Molecular Docking Predicts Complex Structures and Drives Drug Discovery
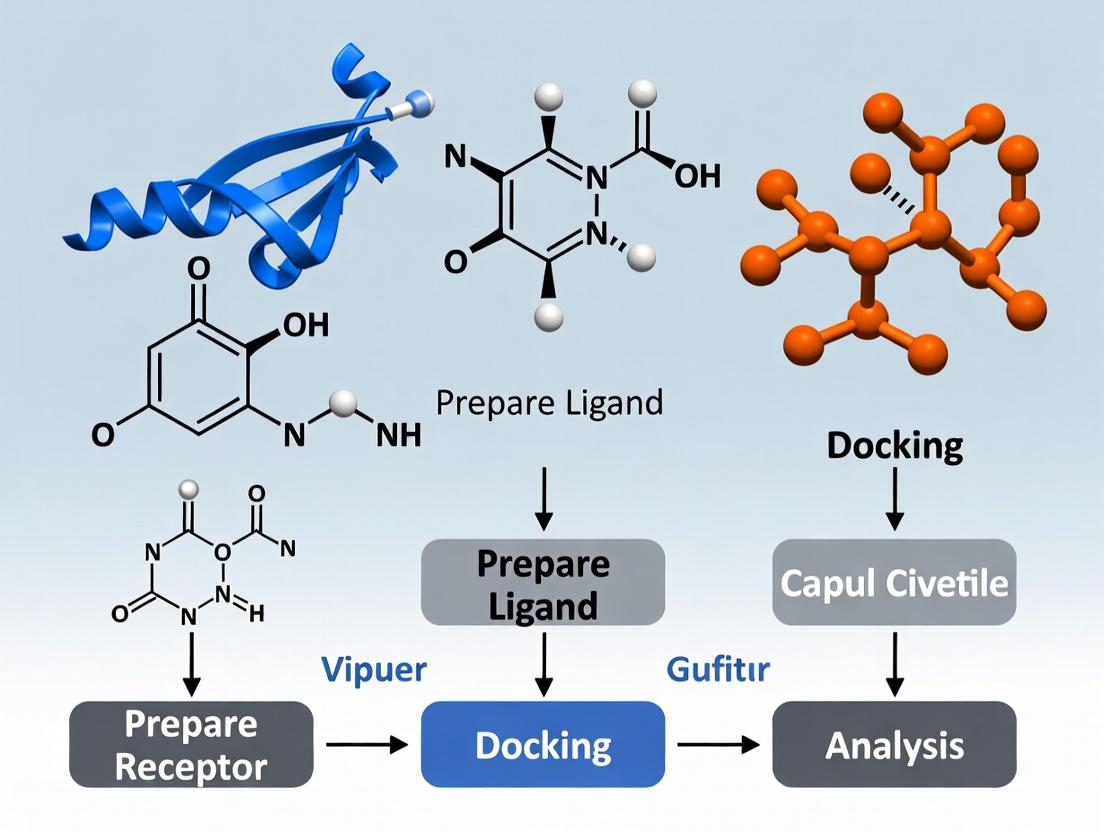
Molecular docking is a cornerstone computational technique in structure-based drug design that predicts the three-dimensional structure of a protein-ligand complex and estimates the binding affinity.
Decoding Protein-Ligand Interactions: How Molecular Docking Predicts Complex Structures and Drives Drug Discovery
Abstract
Molecular docking is a cornerstone computational technique in structure-based drug design that predicts the three-dimensional structure of a protein-ligand complex and estimates the binding affinity. This article provides a comprehensive overview for researchers and drug development professionals, covering the foundational biophysical principles of non-covalent interactions and molecular recognition models. It details core methodological approaches, including search algorithms, scoring functions, and leading software applications in virtual screening and lead optimization. The discussion addresses common challenges such as accounting for protein flexibility and scoring function limitations, while highlighting advanced optimization strategies involving artificial intelligence and molecular dynamics. Finally, the article examines rigorous validation protocols, comparative performance of emerging AI-driven methods against traditional tools, and future directions for integrating computational predictions with experimental validation to accelerate biomedical research.
The Biophysical Blueprint: Core Principles of Protein-Ligand Interactions and Docking Fundamentals
Molecular Docking's Role in Modern Computer-Aided Drug Design (CADD)
Molecular docking is a cornerstone computational technique within Computer-Aided Drug Design (CADD), simulating the prediction of the preferred orientation of a small molecule (ligand) when bound to a target protein. This in-depth guide frames docking within the thesis of how these simulations predict protein-ligand complex structures, which is fundamental to understanding molecular recognition, virtual screening, and lead optimization in modern drug discovery.
Core Principles and Predictive Thesis
The central thesis of molecular docking research posits that the three-dimensional structure of a protein-ligand complex can be predicted by computationally sampling ligand conformations and orientations within the protein's binding site, scoring each pose to estimate binding affinity. This process relies on two core components: a search algorithm and a scoring function.
- Search Algorithm: Explores the conformational and orientational space of the ligand relative to the protein (e.g., systematic, stochastic, or deterministic methods).
- Scoring Function: Quantifies the protein-ligand interaction energy for each generated pose, approximating the binding free energy (ΔG). Types include force-field-based, empirical, and knowledge-based functions.
The accuracy of this prediction is validated by comparing computational models to experimentally determined structures from X-ray crystallography or Cryo-EM.
Table 1: Common Scoring Functions and Their Characteristics
| Scoring Function Type | Basis | Speed | Typical Correlation (R²) with Experimental ΔG | Example Software |
|---|---|---|---|---|
| Force-Field Based | Molecular mechanics terms (van der Waals, electrostatics) | Medium | 0.40 - 0.60 | AutoDock, GOLD |
| Empirical | Weighted sum of interaction terms fit to experimental data | Fast | 0.50 - 0.70 | Glide, ChemScore |
| Knowledge-Based | Statistical preferences from structural databases | Fast | 0.40 - 0.65 | PMF, DrugScore |
| Machine Learning | Trained on structural and affinity data | Varies | 0.60 - 0.80* | RF-Score, NNScore |
* Recent advances show improved performance on specific target classes.
Table 2: Performance Metrics of Docking Programs in Benchmark Studies (CASF)
| Program | Top-Scoring Pose RMSD < 2.0 Å (%) | Scoring Power (Pearson R) | Docking Power (Success Rate) |
|---|---|---|---|
| AutoDock Vina | ~70-80% | 0.60 - 0.65 | ~75% |
| Glide (SP) | ~80-85% | 0.65 - 0.70 | ~80% |
| GOLD | ~75-82% | 0.55 - 0.65 | ~78% |
| Surflex-Dock | ~78-83% | 0.60 - 0.68 | ~77% |
Note: Performance varies significantly with target protein class and ligand properties. Data sourced from recent CASF benchmarks and literature reviews.
Detailed Experimental Protocols
Protocol 1: Standard Molecular Docking Workflow for Virtual Screening
Target Preparation:
- Obtain the 3D structure of the target protein from the PDB (e.g., 7TVP for KRAS G12C).
- Remove water molecules and co-crystallized ligands, except crucial structural waters.
- Add hydrogen atoms, assign protonation states (using tools like PROPKA), and optimize hydrogen bonding networks.
- Define the binding site using the native ligand's coordinates or a predicted active site (e.g., using GRID, SITEMAP).
Ligand Library Preparation:
- Generate a library of 3D small molecule structures in a suitable format (e.g., SDF, MOL2).
- Perform ligand energy minimization using molecular mechanics (MMFF94, GAFF).
- Generate possible tautomers and stereoisomers at physiological pH.
Docking Execution:
- Select a docking program and scoring function (e.g., AutoDock Vina with its default scoring).
- Set search parameters: grid box size centered on the binding site, exhaustiveness.
- Execute docking, generating multiple poses (e.g., 20) per ligand.
Post-Docking Analysis:
- Cluster poses based on RMSD.
- Visually inspect top-ranked poses for key interactions (H-bonds, pi-stacking, hydrophobic contacts).
- Apply post-processing: MM/GBSA rescoring or interaction fingerprint analysis.
Protocol 2: Protocol for Docking Validation (Re-docking/Cross-Docking)
- Complex Selection: Curate a set of high-resolution protein-ligand complexes from the PDB.
- Ligand Extraction: Separate the ligand from the protein structure.
- Re-docking: Dock the extracted ligand back into its original prepared protein structure.
- Pose Comparison: Calculate the Root-Mean-Square Deviation (RMSD) between the top-scoring docked pose and the experimentally determined co-crystallized pose.
- Success Criteria: A docking is considered successful if the heavy-atom RMSD is ≤ 2.0 Å, indicating the method can reproduce the known binding mode.
Visualization of Key Concepts
Molecular Docking Computational Workflow
Taxonomy of Scoring Functions in Docking
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Reagents and Computational Tools for Molecular Docking Studies
| Item/Category | Function in Research | Example Software/Resource |
|---|---|---|
| Protein Structure Repository | Source of experimentally determined target structures. | Protein Data Bank (PDB), AlphaFold DB |
| Small Molecule Database | Source of compounds for virtual screening. | ZINC, ChEMBL, PubChem |
| Molecular Visualization Software | Critical for structure preparation, analysis, and result interpretation. | PyMOL, UCSF Chimera, Maestro |
| Docking Suite | Core software for performing the docking simulation. | AutoDock Vina, Glide (Schrödinger), GOLD (CCDC) |
| Force Field Parameters | Defines atomic partial charges, bond parameters for energy calculations. | CHARMM, AMBER, GAFF |
| Molecular Dynamics (MD) Software | Used for post-docking refinement and stability assessment (MM/GBSA). | GROMACS, AMBER, NAMD |
| High-Performance Computing (HPC) Cluster | Provides computational power for large-scale virtual screens. | Local clusters, Cloud computing (AWS, Azure) |
| Benchmarking Datasets | Standardized sets for validating and comparing docking protocols. | CASF (Comparative Assessment of Scoring Functions), DUD-E |
Within the broader thesis on how molecular docking predicts protein-ligand complex structures, the accurate quantification of non-covalent interactions is paramount. Molecular docking algorithms are computational tools that predict the preferred orientation (pose) and binding affinity of a small molecule (ligand) when bound to a target protein. The predictive power of these tools is fundamentally dependent on the scoring functions that approximate the free energy of binding (ΔG_bind). These scoring functions are mathematical models built upon the physical chemistry of the key non-covalent forces that govern molecular recognition. This guide provides an in-depth technical analysis of these forces, their quantitative characterization, and their integration into docking protocols.
Core Non-Covalent Interactions: Energetics and Characteristics
The stability of a protein-ligand complex arises from the interplay of several non-covalent interactions, each with distinct energetic, geometric, and distance-dependent properties. The following table summarizes key quantitative parameters for these interactions.
Table 1: Quantitative Parameters of Key Non-Covalent Interactions
| Interaction Type | Energy Range (kJ/mol) | Typical Distance Dependence | Directionality | Key Contributors to ΔG |
|---|---|---|---|---|
| Electrostatic (Ion-Ion) | -250 to -20 | 1/r | Low (spherical) | Coulomb's law, desolvation penalty |
| Hydrogen Bond (H-bond) | -40 to -15 | ~1/r⁴ | High (angle/donor-acceptor) | Electrostatics, partial charge transfer |
| Van der Waals (vdW) | -5 to -0.5 | 1/r⁶ (attraction) | Low | Induced dipole fluctuations (London dispersion) |
| Hydrophobic Effect | ~ -0.3 per Ų | N/A | N/A | Entropy gain from released ordered water |
| π-π Stacking | -10 to -5 | Variable (offset preferred) | Moderate | Electrostatics, dispersion |
| Cation-π | -20 to -10 | 1/r⁴ | Moderate | Electrostatics, polarization, dispersion |
| Halogen Bond | -30 to -10 | ~1/r⁴ | High (R–X···O/N angle ~180°) | σ-hole electrostatics, dispersion |
Experimental Protocols for Characterizing Interactions
Understanding these interactions relies on robust experimental techniques.
Protocol 3.1: Isothermal Titration Calorimetry (ITC) for Binding Thermodynamics Objective: To directly measure the binding affinity (K_d), stoichiometry (n), enthalpy change (ΔH), and entropy change (ΔS). Methodology:
- Sample Preparation: Purified protein and ligand are dialyzed into identical, degassed buffer to match chemical potentials.
- Instrument Setup: The cell (~1.4 mL) is filled with protein solution. The syringe is loaded with ligand solution.
- Titration: The ligand is injected in a series of small aliquots (e.g., 2-10 µL) into the stirred cell at constant temperature.
- Heat Measurement: After each injection, the instrument measures the heat (µJ) required to maintain the cell at the same temperature as a reference cell.
- Data Analysis: The integrated heat peaks per injection are fit to a binding model (e.g., one-site) using non-linear least squares regression to extract Kd (ΔG = -RTlnKd), ΔH, and n. ΔS is calculated (ΔG = ΔH - TΔS).
Protocol 3.2: X-ray Crystallography for Structural Characterization Objective: To obtain a high-resolution (<2.0 Å) three-dimensional structure of the protein-ligand complex, visualizing interaction geometries. Methodology:
- Co-crystallization/Soaking: The protein is crystallized in the presence of the ligand (co-crystallization) or ligand is diffused into pre-formed protein crystals (soaking).
- Data Collection: Crystals are flash-cooled. X-rays are diffracted by the crystal, producing a pattern recorded on a detector.
- Phase Determination: Phases are solved via molecular replacement (using a known homologous structure) or experimental phasing.
- Model Building & Refinement: An atomic model is built into the electron density map. The model (including ligand pose) is iteratively refined against the diffraction data (R-factors). Hydrogen bonds and vdW contacts are measured using software like PyMOL or CCP4.
Protocol 3.3: Surface Plasmon Resonance (SPR) for Kinetics Objective: To measure the real-time association (kon) and dissociation (koff) rate constants, from which Kd (koff/k_on) is derived. Methodology:
- Immobilization: The target protein is covalently immobilized on a dextran-coated gold sensor chip.
- Ligand Flow: Ligand solutions at varying concentrations are flowed over the chip surface in a continuous buffer stream.
- Signal Detection: Binding changes the refractive index at the chip surface, detected as a shift in resonance angle (Response Units, RU).
- Kinetic Analysis: The association and dissociation phases of the sensorgram are globally fit to a 1:1 Langmuir binding model to extract kon and koff.
Visualization of Molecular Docking Workflow and Energy Components
The following diagram illustrates the standard molecular docking workflow and how non-covalent interactions are integrated into scoring functions.
Title: Molecular Docking Workflow and Energy Scoring
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Protein-Ligand Binding Studies
| Item | Function/Application | Key Consideration |
|---|---|---|
| High-Purity Recombinant Protein | The target for biophysical assays (ITC, SPR). | Requires homogeneous, monodisperse, functional protein. Systems: E. coli, insect, mammalian. |
| Analytical Grade Ligands | Small molecule compounds for binding studies. | Must be >95% pure, solubilized in compatible buffer (DMSO stock common). |
| ITC Buffer Kit | Pre-formulated, matched buffer salts for ITC. | Minimizes heats of dilution; critical for accurate ΔH measurement. |
| SPR Sensor Chip (CM5) | Gold sensor chip with carboxymethylated dextran matrix. | Common for amine-coupling immobilization of proteins. |
| Crystallization Screening Kit | Sparse matrix of chemical conditions for crystal growth. | Commercial screens (e.g., from Hampton Research) sample diverse precipitant, pH, salt space. |
| Cryoprotectant (e.g., Glycerol, PEG) | Protects crystals during flash-cooling for X-ray data collection. | Prevents ice formation that destroys crystal order. |
| Analysis Software (PyMOL, MOE, Schrodinger) | Visualizes structures, measures distances/angles, analyzes binding sites. | Essential for interpreting structural data and docking results. |
| Docking Software (AutoDock Vina, Glide, GOLD) | Performs the computational pose prediction and scoring. | Choice depends on scoring function, speed, and user expertise. |
Molecular docking is a pivotal computational technique in structural biology and drug discovery, aiming to predict the three-dimensional structure of a protein-ligand complex. The accuracy and predictive power of docking algorithms are fundamentally governed by the underlying model of molecular recognition they employ. This whitepaper examines the evolution from the classic Lock-and-Key paradigm to the more dynamic Conformational Selection and Induced Fit models. Understanding these biophysical principles is critical for developing and validating docking protocols, as they inform scoring functions, search algorithms, and the treatment of protein flexibility—a major challenge in accurately predicting binding poses and affinities.
Evolution of Recognition Models
Lock-and-Key Model
Proposed by Emil Fischer in 1894, this model posits a rigid, pre-existing complementarity between the ligand (key) and the protein's binding site (lock). It assumes minimal conformational change upon binding.
Relevance to Docking: Early rigid-body docking algorithms were based on this model, treating both receptor and ligand as static shapes. While computationally efficient, this approach often fails for flexible systems.
Induced Fit Model
Proposed by Daniel Koshland in 1958, this model asserts that the binding site is not perfectly complementary to the ligand. The ligand induces a conformational change in the protein to achieve optimal binding.
Relevance to Docking: Modern docking software incorporates aspects of induced fit through methods like side-chain flexibility, protein ensemble docking, or on-the-fly minimization during the docking search.
Conformational Selection Model
This contemporary model, gaining prominence in the early 2000s, proposes that the protein exists in an equilibrium of multiple pre-existing conformations. The ligand selectively binds to and stabilizes a specific, complementary conformation, shifting the population equilibrium.
Relevance to Docking: This is the conceptual foundation for advanced techniques like ensemble docking, where a ligand is docked against a collection of protein conformations derived from molecular dynamics (MD) simulations, NMR, or multiple crystal structures.
Quantitative Comparison of Model Characteristics
Table 1: Comparative Analysis of Molecular Recognition Models
| Feature | Lock-and-Key | Induced Fit | Conformational Selection |
|---|---|---|---|
| Protein State | Single, rigid conformation. | Flexible, adapts upon ligand encounter. | Ensemble of pre-existing conformations. |
| Ligand Role | Passive key. | Inducer of change. | Selective stabilizer. |
| Binding Kinetics | Often described as a single-step process. | Two-step: encounter followed by adaptation. | Ligand binds to a rare pre-existing state, shifting equilibrium. |
| Key Experimental Evidence | X-ray structures of apo/holo proteins with identical site geometry. | X-ray structures showing significant backbone/sidechain movement between apo/holo forms. | NMR relaxation dispersion, single-molecule FRET, kinetic studies showing multi-state equilibria. |
| Computational Docking Approach | Rigid-body docking. | Flexible docking, protein minimization. | Ensemble docking, MD-based sampling. |
| Primary Limitation | Neglects protein dynamics and flexibility. | May overemphasize ligand-induced changes over pre-existing populations. | Requires extensive sampling of protein conformational space. |
Key Experimental Methodologies
Isothermal Titration Calorimetry (ITC) for Binding Thermodynamics
Protocol: A solution of the protein is placed in the sample cell. A syringe loaded with a concentrated ligand solution titrates the protein. After each injection, the power required to maintain the sample cell at the same temperature as the reference cell (filled with buffer) is measured. Data Output: Direct measurement of binding constant (Kd), enthalpy change (ΔH), and stoichiometry (n). Entropy (ΔS) is calculated. A negative ΔH and positive ΔS suggest a binding event driven by both specific interactions and increased disorder (e.g., release of ordered water).
Nuclear Magnetic Resonance (NMR) Spectroscopy for Detecting Dynamics
Protocol (Cα Chemical Shift Perturbation & Relaxation Dispersion):
- Prepare 15N- or 13C-labeled protein samples in both apo and ligand-bound states.
- Acquire 2D 1H-15N HSQC spectra. Chemical shift changes in backbone amides indicate regions affected by binding.
- For relaxation dispersion (Carr-Purcell-Meiboom-Gill, CPMG), measure R2 (transverse relaxation rate) as a function of applied pulse frequency (νCPMG). A dependence of R2 on νCPMG reveals conformational exchange on the μs-ms timescale. Interpretation: Conformational selection is supported if minor states detected in the apo protein spectrum correspond to the ligand-bound conformation.
X-ray Crystallography for Structural Snapshots
Protocol:
- Co-crystallize the protein with the ligand or soak the ligand into pre-formed apo protein crystals.
- Collect X-ray diffraction data at a synchrotron source.
- Solve the structure by molecular replacement (using a known homologous structure) and refine. Interpretation: Comparison of apo and holo structures provides static snapshots. Multiple, distinct conformations of a binding site in different crystal forms (polymorphism) can be evidence for a conformational ensemble.
Molecular Dynamics (MD) Simulations for Sampling Conformations
Protocol (Ensemble Generation for Docking):
- Start with an apo protein structure in an explicit solvent (water/ions) box.
- Energy-minimize the system, then equilibrate under constant temperature (NVT) and pressure (NPT) conditions.
- Run a production MD simulation (nanoseconds to microseconds). Save trajectory frames at regular intervals (e.g., every 100 ps).
- Cluster the saved frames based on binding site geometry (e.g., RMSD) to select a representative ensemble of distinct conformations for subsequent ensemble docking.
Visualizing Concepts and Workflows
Diagram 1: Lock-and-Key vs. Conformational Selection
Diagram 2: Ensemble Docking Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Studying Molecular Recognition
| Item | Function in Research | Example/Note |
|---|---|---|
| Recombinant Protein Expression Systems | Produce pure, homogenous protein for biophysical assays. | E. coli, insect cell (baculovirus), or mammalian (HEK293) systems. Isotopic labeling (15N, 13C) for NMR. |
| Thermal Shift Dye (e.g., SYPRO Orange) | High-throughput screening of ligand binding by monitoring protein thermal stability (DSF). | Binding often stabilizes protein, increasing melting temperature (Tm). |
| Surface Plasmon Resonance (SPR) Chips | Immobilize protein to measure real-time binding kinetics (kon, koff) of ligands in flow. | CM5 dextran chips (carboxylated) for amine coupling. |
| Crystallization Screening Kits | Identify initial conditions for growing protein/co-crystals. | Sparse matrix screens (e.g., from Hampton Research, Jena Bioscience). |
| NMR Buffer Kits | Prepare deuterated, pH-adjusted buffers compatible with NMR spectroscopy. | Minimizes interfering signals and maintains protein activity. |
| Molecular Dynamics Software | Simulate protein motion and generate conformational ensembles. | GROMACS, AMBER, NAMD, CHARMM. |
| Docking Software Suites | Computationally predict binding poses and scores. | AutoDock Vina, Glide (Schrödinger), GOLD (CCDC), Rosetta. |
| Fluorescently Labeled Ligands/Proteins | Enable binding studies via fluorescence anisotropy (FA) or Förster Resonance Energy Transfer (FRET). | Requires site-specific labeling (e.g., via cysteine chemistry). |
Molecular docking is a computational technique at the heart of structure-based drug design, predicting the three-dimensional structure of a protein-ligand complex. Its accuracy and predictive power are wholly dependent on the synergistic operation of two core components: the conformational search algorithm, which explores the vast landscape of possible ligand orientations and conformations within the binding site, and the scoring function, which evaluates and ranks these poses to identify the most likely binding mode. This article, framed within a broader thesis on how molecular docking predicts complex structures, details the technical intricacies of these two engines and their integration.
Conformational Search: Sampling the Possibility Space
The first challenge is to efficiently sample the astronomical number of possible ligand poses. Current methodologies balance computational cost with coverage.
Key Search Algorithms & Protocols
1. Systematic Search (Exhaustive):
- Protocol: The ligand's rotatable bonds are rotated through a user-defined grid of angles (e.g., every 10 or 30 degrees). All combinations of these rotations are generated and evaluated. This method is thorough but computationally intractable for ligands with more than ~10 rotatable bonds.
- Typical Implementation: Used in early docking programs (e.g., DOCK 3.x) and often combined with rigid protein approximations.
2. Stochastic/Monte Carlo Methods:
- Protocol: A starting pose is randomly perturbed by translating, rotating, or rotating around bonds. The new pose is accepted or rejected based on the Metropolis criterion (accepting poses with better scores, and sometimes worse scores with a probability that decreases over "temperature" cycles). This process is repeated for thousands to millions of iterations.
- Protocol Detail (Basic Cycle):
- Generate initial ligand pose at random within the binding site.
- Randomly translate (±0.5 Å), rotate (±30°), or rotate a torsion (±30°).
- Score the new pose.
- If score improves, accept the pose. If score worsens, accept with probability P = exp(-ΔScore / kT), where T is a "simulated temperature" parameter that decreases over the simulation (simulated annealing).
- Repeat from step 2.
3. Genetic Algorithms (GA):
- Protocol: Poses ("individuals") are encoded as chromosomes representing their position, orientation, and torsion angles. A population of poses undergoes "evolution" via selection (based on score), crossover (combining parts of two parent poses), and mutation (random changes). Over generations, the population converges toward an optimal pose.
- Protocol Detail (Generation Cycle):
- Initialize a population of N random poses (e.g., N=50).
- Score all individuals.
- Select top-ranked individuals as "parents."
- Create "offspring" by crossover (e.g., mixing torsions from two parents) and mutation (randomly altering a gene).
- Form a new population from parents and offspring.
- Repeat from step 2 for G generations (e.g., G=1000).
4. Molecular Dynamics (MD)-Based Sampling:
- Protocol: Newton's equations of motion are solved for the ligand (and sometimes flexible protein residues) within the binding site, using a force field. This generates a time-evolving trajectory of poses. While highly accurate, plain MD is computationally expensive for broad sampling. Accelerated MD (aMD) or Steered MD (sMD) apply biases to overcome energy barriers more efficiently.
Quantitative Comparison of Search Methods
Table 1: Comparison of Core Conformational Search Algorithms
| Method | Sampling Nature | Computational Cost | Strengths | Weaknesses | Common Software |
|---|---|---|---|---|---|
| Systematic | Deterministic, Exhaustive | Very High (Exponential) | Guaranteed local completeness | Combinatorial explosion, impractical for flexible ligands | Early DOCK, FRED |
| Stochastic (MC) | Random, Non-deterministic | Moderate to High | Can escape local minima, good for flexible ligands | No completeness guarantee; results may vary between runs | AutoDock, ICM |
| Genetic Algorithm | Population-based, Evolutionary | Moderate | Efficient global search, good parallelism | Parameter-dependent, may converge prematurely | AutoDock Vina, GOLD |
| MD-Based | Physics-based, Deterministic | Very High | High accuracy, includes explicit dynamics | Extremely resource-intensive for sampling | AMBER, NAMD, Desmond |
Docking Workflow: Search and Score
Scoring Functions: The Discriminatory Judge
Scoring functions mathematically approximate the binding free energy (ΔG_bind) to distinguish near-native poses from decoys. They fall into three primary categories.
Types of Scoring Functions
1. Force Field-Based:
- Methodology: Calculate ΔG using molecular mechanics terms (van der Waals, electrostatic) and an implicit solvation model. The binding energy is: ΔGbind = Ecomplex - (Eprotein + Eligand).
- Protocol: After generating a pose, calculate the energy using a force field (e.g., AMBER, CHARMM). Protein-ligand electrostatic interactions are often calculated using a pre-computed grid to speed up evaluation.
2. Empirical:
- Methodology: Fit a linear equation to experimental binding affinity data using descriptors (e.g., hydrogen bonds, hydrophobic contacts, rotatable bond penalty). ΔGbind = Σ (ci * Di), where ci are fitted coefficients and D_i are feature counts.
- Protocol: For a given pose, the software counts interaction features (e.g., number of H-bonds, metal contacts, buried surface area). These counts are multiplied by pre-trained coefficients and summed to yield a score.
3. Knowledge-Based:
- Methodology: Derive potentials of mean force from statistical analysis of atom-pair frequencies in known protein-ligand complexes (PDB). The score is Σ Aij(r) = -kB T ln [fobsij(r) / frefij(r)], where f is the observed frequency of atom pair ij at distance r.
- Protocol: A distance histogram is created for all atom pairs (C-C, C-N, O-H, etc.) from a large database of complexes. For a new pose, the distance-dependent score for each atom pair is looked up and summed.
Quantitative Comparison of Scoring Functions
Table 2: Comparison of Core Scoring Function Types
| Type | Theoretical Basis | Speed | Typical Correlation (R²) with Exp. ΔG* | Key Strengths | Key Limitations |
|---|---|---|---|---|---|
| Force Field | Physics (MM potentials) | Moderate | 0.40 - 0.55 | Physically intuitive, good for pose ranking | Sensitive to parameterization, neglects entropy |
| Empirical | Linear regression on data | Very Fast | 0.50 - 0.65 | Fast, optimized for affinity prediction | Training-set dependent, risk of overfitting |
| Knowledge-Based | Statistics of known structures | Fast | 0.45 - 0.60 | No training data needed, captures implicit effects | Interpretability issues, database bias |
| Machine Learning | Non-linear models on features | Fast (after training) | 0.60 - 0.80+ | High predictive accuracy for affinity | Black-box nature, heavy training data dependence |
Note: R² values are approximate ranges from recent benchmarks (e.g., PDBbind, CASF). Machine Learning-based functions (e.g., RF-Score, Δvina XGB) now often lead in affinity prediction.
Scoring Function Evaluation Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Resources for Molecular Docking
| Item / Resource | Category | Function / Purpose | Example (Vendor/Provider) |
|---|---|---|---|
| Protein Data Bank (PDB) | Data Source | Repository of experimentally solved 3D protein structures, essential for obtaining target coordinates. | www.rcsb.org |
| Ligand Preparation Tool | Software | Processes 2D ligand structures (e.g., SDF) into 3D, assigns protonation states, and generates low-energy conformers. | OpenBabel, LigPrep (Schrödinger), MOE |
| Protein Preparation Suite | Software | Prepares protein structure: adds hydrogens, optimizes H-bond networks, fixes missing residues/side chains. | Protein Preparation Wizard (Schrödinger), UCSF Chimera, BIOVIA Discovery Studio |
| Docking Software Suite | Core Engine | Integrates search algorithms and scoring functions to perform the docking simulation. | AutoDock Vina, GOLD, Glide (Schrödinger), DOCK 6 |
| Scoring Function Library | Software/Algorithm | Provides diverse scoring functions for pose ranking or re-scoring to improve prediction accuracy. | Smina (Vina variant), RF-Score, NNScore, DSX |
| Molecular Visualization System | Analysis Tool | Visualizes docking results, analyzes protein-ligand interactions (H-bonds, hydrophobic surfaces). | PyMOL, UCSF Chimera, Maestro (Schrödinger) |
| Benchmarking Dataset | Validation | Curated sets of protein-ligand complexes with known structures/affinities for method validation. | PDBbind, CASF (Comparative Assessment of Scoring Functions), DUD-E (Decoys) |
| High-Performance Computing (HPC) Cluster | Infrastructure | Provides the computational power needed for large-scale virtual screening or MD-based docking. | Local university cluster, Cloud (AWS, Azure), Google Cloud Platform |
From Algorithm to Action: Docking Strategies, Software, and Practical Applications in Drug Discovery
This whitepaper provides an in-depth technical examination of core molecular docking methodologies, framed within the broader thesis research question: How does molecular docking predict protein-ligand complex structures? Docking is a computational cornerstone in structural biology and drug discovery, aiming to predict the preferred orientation (pose) and binding affinity (score) of a small molecule (ligand) when bound to a target macromolecule (receptor, typically a protein). The accuracy of these predictions is fundamentally constrained by the treatment of molecular flexibility, leading to the evolution of three primary strategies: Rigid, Flexible, and Ensemble Docking.
Core Docking Methodologies
Rigid Body Docking
Concept: Treats both the protein receptor and the ligand as rigid, unchanging structures. The search algorithm explores only the translational and rotational degrees of freedom of the ligand relative to the protein binding site. Thesis Context Application: Serves as a foundational model. Its performance benchmarks the necessity for incorporating flexibility, as it fails when induced fit or conformational selection mechanisms are significant. Typical Algorithms: Fast Fourier Transform (FFT) correlation approaches (e.g., ZDOCK, GRAMM). Best For: Preliminary screening of ligands against static, well-defined binding pockets with minimal expected side-chain movement.
Flexible Ligand Docking
Concept: The protein receptor remains rigid, but the ligand is allowed full conformational flexibility (rotatable bonds). This is the most common standard in modern docking. Thesis Context Application: Addresses a key variable—ligand conformation—acknowledging that ligands adopt different shapes in solution versus the bound state. Typical Algorithms: Stochastic methods (Genetic Algorithms, Monte Carlo), systematic search (incremental construction), or molecular dynamics-based methods. Best For: Virtual screening (VS) and lead optimization where ligand flexibility is critical but the protein target is considered stable.
Flexible & Ensemble Docking
- Flexible Side-Chain Docking: Allows specific protein side-chains in the binding site to rotate during the docking simulation.
- Ensemble Docking: Uses multiple protein receptor structures (an ensemble) derived from NMR models, molecular dynamics (MD) simulation snapshots, or alternative crystal structures. Docking is performed against each member of the ensemble, and results are aggregated. Thesis Context Application: Directly engages with the protein flexibility problem, a major challenge in accurate prediction. It tests the hypotheses of induced fit (ligand causes protein change) and conceptual selection (ligand selects pre-existing protein conformation). Best For: Targets with highly flexible binding sites, allosteric sites, or where significant conformational changes upon binding are known or suspected.
Quantitative Performance Comparison
The following table summarizes key performance metrics and characteristics of the three methodologies, based on recent benchmarking studies (e.g., DUD-E, DEKOIS 2.0).
Table 1: Comparative Analysis of Docking Methodologies
| Metric / Characteristic | Rigid Docking | Flexible Ligand Docking | Ensemble Docking |
|---|---|---|---|
| Computational Speed | Very Fast (seconds/pose) | Moderate (seconds-minutes/pose) | Slow (minutes-hours/ligand)* |
| Typical Pose RMSD Accuracy | >2.5 Å (for flexible targets) | 1.5 - 2.5 Å | 1.0 - 2.0 Å (for matched conformers) |
| Enrichment Factor (EF₁%) in VS | Low | Moderate to High | Highest (when ensemble is representative) |
| Handles Protein Flexibility | No | No | Yes |
| Primary Search Degrees of Freedom | 6 (Rotation + Translation) | 6 + N (N=rotatable bonds) | 6 + N + M (M=protein torsions) |
| Key Limitation | Neglects biological flexibility | Neglects protein flexibility | Ensemble generation & selection bias |
| Representative Software | ZDOCK, GRAMM-X | AutoDock Vina, Glide, GOLD | Schrödinger IFD, AutoDockFR, RosettaDock |
*Speed depends on ensemble size.
Detailed Experimental Protocols
Protocol for Standard Flexible Ligand Docking (Using AutoDock Vina)
This protocol is a benchmark for thesis research into pose prediction accuracy.
System Preparation:
- Protein: Obtain PDB structure. Remove water molecules, cofactors, and heteroatoms. Add polar hydrogens and assign Gasteiger charges using tools like MGLTools or UCSF Chimera.
- Ligand: Obtain ligand structure in 2D/3D format (e.g., SDF). Convert to PDBQT format, defining rotatable bonds and root.
Grid Box Definition:
- Identify the binding site centroid (from crystallographic ligand or literature).
- Define a 3D search space (grid box) centered on this centroid. Typical size: 20x20x20 Å or larger to fully encompass the site. This box is where the ligand will be placed and searched.
Configuration File Creation:
- Create a configuration file (
config.txt) specifying:receptor = protein.pdbqtligand = ligand.pdbqtcenter_x, center_y, center_z = [coordinates]size_x, size_y, size_z = [dimensions]exhaustiveness = 8(default, can be increased for accuracy).num_modes = 9(number of output poses).
- Create a configuration file (
Docking Execution:
- Run the command:
vina --config config.txt --log log.txt
- Run the command:
Post-Processing & Analysis:
- Analyze the output file (
ligand_out.pdbqt) containing ranked poses. - Calculate Root-Mean-Square Deviation (RMSD) of predicted poses versus a known crystallographic pose to evaluate accuracy.
- Examine binding scores (in kcal/mol) for relative affinity ranking.
- Analyze the output file (
Protocol for Ensemble Docking (Generic Workflow)
This protocol tests the "conformational selection" hypothesis within the thesis.
Ensemble Generation:
- Source 1 (Experimental): Collect multiple experimental structures (X-ray, NMR) of the target from the PDB.
- Source 2 (Computational): Perform a Molecular Dynamics (MD) simulation of the apo (unbound) protein. Extract snapshots at regular intervals (e.g., every 10 ns) to capture conformational diversity.
Ensemble Pre-processing & Alignment:
- Superimpose all protein structures onto a common reference frame (e.g., the backbone of the protein core) to ensure the binding site coordinates are comparable.
Docking Against Ensemble:
- Perform flexible ligand docking (as in Protocol 4.1) of the ligand against each individual protein conformation in the ensemble.
Result Aggregation & Consensus Scoring:
- Collect all predicted poses and scores from each docking run.
- Consensus Method 1: Rank ligands by their best score across any ensemble member.
- Consensus Method 2: Cluster similar poses across the ensemble and rank by average score or cluster population.
- Analyze which protein conformations yielded the best-scoring poses, providing insight into the likely selected binding state.
Visualization of Docking Workflows and Relationships
Title: Decision Workflow for Selecting a Docking Methodology
Title: Ensemble Docking Protocol Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools and Resources for Molecular Docking Research
| Item / Resource | Category | Function / Purpose |
|---|---|---|
| RCSB Protein Data Bank (PDB) | Database | Primary repository for experimentally determined 3D structures of proteins and nucleic acids. Source of receptor and complex structures for validation. |
| ZINC20 Database | Database | Curated commercial library of over 200 million purchasable compounds in ready-to-dock 3D formats. Essential for virtual screening. |
| AutoDock Vina | Software | Widely-used, open-source docking program offering a good balance of speed and accuracy for flexible ligand docking. |
| Schrödinger Suite (Glide) | Software | Commercial, industry-standard platform offering high-accuracy docking (Glide), induced fit docking (IFD), and extensive MD capabilities. |
| GROMACS | Software | High-performance, open-source MD software package for generating conformational ensembles via molecular dynamics simulations. |
| PyMOL / UCSF Chimera | Software | Visualization tools critical for preparing structures, analyzing docking poses (RMSD, interactions), and creating publication-quality figures. |
| Python (with RDKit, MDAnalysis) | Programming/API | Enables automation of docking pipelines, custom analysis, and the integration of machine learning approaches. |
| DUD-E / DEKOIS 2.0 | Benchmark Set | Curated datasets for benchmarking docking methods, containing active molecules and decoys to assess enrichment. |
| High-Performance Computing (HPC) Cluster | Hardware | Essential for computationally intensive tasks like ensemble docking, large-scale virtual screens, and MD simulations. |
Molecular docking is a cornerstone computational technique in structural bioinformatics and computer-aided drug design (CADD). Within the broader thesis of predicting protein-ligand complex structures, docking tools serve as the primary engines for simulating and scoring the binding of a small molecule (ligand) within a protein's active site. The accuracy of these predictions is critical for understanding molecular recognition, elucidating biological mechanisms, and accelerating drug discovery by identifying and optimizing potential lead compounds. This overview provides a technical comparison of widely used docking software, detailing their methodologies, scoring functions, and experimental protocols.
Core Docking Algorithms and Scoring Functions
The predictive power of a docking tool hinges on its search algorithm and scoring function. The search algorithm explores the conformational and orientational space of the ligand within the binding site, while the scoring function evaluates and ranks the predicted poses.
Search Algorithms
- Systematic Search: Exhaustively explores torsional degrees of freedom (e.g., Glide's hierarchical filters).
- Stochastic/Monte Carlo: Uses random changes and an acceptance criterion to sample poses (e.g., early AutoDock).
- Genetic Algorithms: Evolves a population of poses using selection, crossover, and mutation (e.g., GOLD).
- Molecular Dynamics (MD)-Based: Uses simulated annealing or gradient-based minimization (e.g., AutoDock Vina).
Scoring Functions
- Force Field-Based: Calculate energy using molecular mechanics terms (van der Waals, electrostatics).
- Empirical: Fit parameters to experimental binding affinity data using linear regression.
- Knowledge-Based: Derive potentials from statistical analysis of atom-pair frequencies in known protein-ligand complexes.
Comparative Analysis of Major Docking Tools
The following table summarizes the key characteristics, algorithms, and typical use cases for prominent docking programs.
Table 1: Comparison of Key Molecular Docking Software
| Tool | Developer | Core Search Algorithm | Primary Scoring Function | License/Cost | Typical Application Context |
|---|---|---|---|---|---|
| AutoDock Vina | The Scripps Research Institute | Iterated Local Search, Monte Carlo | Hybrid (Empirical + Knowledge-based) | Open Source (Apache 2.0) | High-throughput virtual screening, pose prediction. |
| Glide | Schrödinger | Systematic, hierarchical search | Empirical (GlideScore) | Commercial | High-accuracy pose prediction & scoring, lead optimization. |
| GOLD | CCDC | Genetic Algorithm | Empirical (ChemScore, GoldScore) | Commercial | Flexible ligand & side-chain docking, scaffold hopping. |
| AutoDock 4/GPU | Scripps | Lamarckian Genetic Algorithm | Semi-empirical Force Field | Open Source | Detailed binding energy estimation, flexible residues. |
| FRED (OE) | OpenEye | Exhaustive systematic search | Hybrid (Shapegauss, Chemgauss) | Commercial | Ultra-fast high-throughput screening. |
| rDock | University of Hamburg | Stochastic search + MC minimization | Empirical (Rbt) | Open Source (GPL) | Structure-based design, pharmacophore docking. |
| SwissDock | SIB / UNIL | EADock DSS (heuristic) | CHARMM force field | Free Web Server | Easy-access academic research, teaching. |
Table 2: Benchmark Performance Metrics (Representative Data from Recent Evaluations)
| Tool | Average RMSD (<2Å) | Success Rate (Top Pose) | Typical Runtime per Ligand | Key Strength |
|---|---|---|---|---|
| Glide (SP) | 1.2 - 1.5 Å | ~75-80% | 1-3 minutes | Pose accuracy, scoring consistency. |
| GOLD (ChemScore) | 1.3 - 1.7 Å | ~70-78% | 2-5 minutes | Handling ligand flexibility. |
| AutoDock Vina | 1.5 - 2.0 Å | ~65-75% | 1-2 minutes | Speed & accuracy balance, accessibility. |
| AutoDock 4 | 1.8 - 2.5 Å | ~60-70% | 5-15 minutes | Binding free energy estimation. |
Note: Performance is highly dependent on the protein target, ligand set, and preparation protocols. Data is synthesized from recent CASF benchmarks and community assessments.
Experimental Protocol for a Standard Docking Workflow
A robust docking study follows a standardized pipeline to ensure reproducibility and reliability.
Protocol: Standard Molecular Docking and Virtual Screening
Target Protein Preparation:
- Source: Obtain 3D structure from PDB (e.g., 3ERT for estrogen receptor). Prefer high-resolution (<2.0 Å) structures with a bound ligand.
- Processing: Remove water molecules, co-crystallized ligands, and irrelevant ions. Add missing hydrogen atoms and assign protonation states (e.g., using
reduceorEpik). Critical: Determine the protonation state of histidine residues (HID, HIE, HIP) relevant to binding. - Energy Minimization: Perform a restrained minimization (e.g., with
OPLS4orAMBERforce field) to relieve steric clashes, keeping heavy atoms fixed.
Binding Site Definition:
- Define a 3D grid box centered on the known co-crystallized ligand or a predicted active site (e.g., using
SiteMap). Typical box dimensions are 20x20x20 Å with 0.375 Å grid spacing for Vina, or 10 Å padding around the ligand for Glide.
- Define a 3D grid box centered on the known co-crystallized ligand or a predicted active site (e.g., using
Ligand Library Preparation:
- Format: Convert library (e.g., SDF, SMILES) to 3D coordinates.
- Optimization: Generate tautomers, stereoisomers, and protonation states at physiological pH (e.g., with
LigPrep,MOE). - Energy Minimization: Minimize each ligand using an appropriate force field (e.g.,
MMFF94s).
Molecular Docking Execution:
- Software-Specific Commands:
- AutoDock Vina:
vina --receptor protein.pdbqt --ligand ligand.pdbqt --config config.txt --out output.pdbqt - Glide (Schrödinger): Use the
glidemodule via Maestro GUI or command line with an input.infile. - GOLD: Configure the
config.txtfile specifying protein, ligand, genetic algorithm parameters, and scoring function, then executegold_auto.
- AutoDock Vina:
- Software-Specific Commands:
Pose Analysis and Scoring:
- Cluster docked poses by RMSD (e.g., 2.0 Å cutoff).
- Inspect the top-ranked poses for key hydrogen bonds, hydrophobic contacts, and salt bridges.
- Apply post-docking scoring or rescoring with a more rigorous method (e.g., MM-GBSA) to improve affinity ranking.
Validation:
- Re-docking: Dock the native co-crystallized ligand back into the prepared protein. A successful protocol should reproduce the experimental pose with RMSD < 2.0 Å.
- Decoy Set: Use a database of known actives and inactives/decoys to calculate enrichment factors (EF) and ROC curves.
Diagram: Standard Molecular Docking Workflow
Diagram: Classification of Scoring Functions
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Reagents and Materials for Docking & Validation Experiments
| Item | Function / Purpose | Example/Supplier |
|---|---|---|
| Purified Target Protein | Essential for experimental validation (SPR, ITC) of docking hits. Recombinantly expressed and purified protein. | His-tagged kinase expressed in HEK293 cells. |
| Compound Library | Collection of small molecules for virtual & experimental screening. | Enamine REAL Database, MCULE, in-house collections. |
| Co-crystallized Ligand | Reference molecule from PDB structure used for re-docking validation. | Retrieved from PDB file (e.g., HCL from 3ERT). |
| Assay Buffer (e.g., PBS) | For in vitro binding or activity assays to test predicted ligands. | 1X Phosphate Buffered Saline, pH 7.4. |
| Surface Plasmon Resonance (SPR) Chip | For real-time, label-free measurement of binding kinetics (KD, ka, kd). | CMS Series S Chip (Cytiva). |
| ITC Cell & Syringe | For isothermal titration calorimetry to determine binding affinity (KD) and thermodynamics (ΔH, ΔS). | MicroCal Peltier cell (Malvern Panalytical). |
| Crystallization Kits | For structure determination of protein-hit complexes to confirm docking predictions. | Hampton Research Crystal Screens. |
| High-Performance Computing (HPC) Cluster | Computational resource for running large-scale virtual screens and MD simulations. | Local Linux cluster or cloud computing (AWS, Azure). |
This technical guide details a standard computational workflow for protein-ligand molecular docking, framed within the context of broader research on how molecular docking predicts the three-dimensional structure of protein-ligand complexes. This methodology is foundational for structure-based drug design.
Protein Structure Acquisition and Preparation
The initial step involves obtaining a high-quality three-dimensional structure of the target protein, typically from the Protein Data Bank (PDB). The choice of structure is critical; X-ray crystallography structures with high resolution (<2.0 Å) and low R-factors are preferred. Homology models can be used if no experimental structure is available.
Experimental Protocol: Protein Preparation
- Retrieval: Download the PDB file (e.g.,
7sg8.pdb) from the RCSB PDB. - Structure Cleaning: Remove all non-protein entities except essential cofactors or crystallographic waters. Delete alternative conformations (retain the highest occupancy).
- Missing Components: Add missing hydrogen atoms appropriate for the chosen pH (e.g., 7.4). Model missing loops using homology modeling or loop building algorithms.
- Protonation States: Assign correct protonation states to histidine, aspartic acid, glutamic acid, and lysine residues using tools like PROPKA or H++.
- Energy Minimization: Perform a restrained minimization (e.g., using OPLS4 or CHARMM force fields) to relieve steric clashes introduced during hydrogen addition, with a root-mean-square deviation (RMSD) constraint of 0.3 Å on heavy atoms to preserve the experimental conformation.
Diagram Title: Protein Preparation Workflow
Ligand Structure Preparation
Ligand structures can be sourced from small-molecule databases like PubChem or ZINC, or designed de novo. They must be converted into a suitable 3D format with correct chemistry.
Experimental Protocol: Ligand Preparation
- Retrieval/Design: Obtain the ligand's 2D structure (SMILES or SDF).
- 3D Generation: Generate a 3D conformation using energy minimization (e.g., with the MMFF94s force field).
- Tautomerization and Stereochemistry: Enumerate possible tautomers and stereoisomers at the target pH. For virtual screening, all relevant states may be considered.
- Charge Assignment: Assign partial atomic charges using methods like Gasteiger-Marsili or AM1-BCC.
- File Format Conversion: Output the ligand in required formats (e.g., MOL2, SDF, PDBQT).
Binding Site Definition and Grid Generation
The spatial region where docking calculations occur must be defined, typically centered on a known active site or a predicted binding pocket.
Experimental Protocol: Grid Generation
- Site Identification: Use coordinates from a co-crystallized ligand, literature data, or computational pocket detection tools (e.g., FTmap, SiteMap).
- Grid Box Definition: Define a 3D grid box large enough to accommodate the ligand's rotational and translational freedom. Common sizes are 20x20x20 ų or 25x25x25 ų.
- Grid Calculation: Using software like AutoDock Tools or Schrödinger's Glide, pre-calculate energy grids for each atom type in the ligand, evaluating interactions with the protein at every grid point. This maps the protein's energetic landscape.
Molecular Docking Execution
The docking algorithm computationally samples the ligand's conformational, orientational, and positional space within the binding site to identify low-energy binding poses.
Experimental Protocol: Docking with AutoDock Vina
- Configuration: Prepare a configuration file (
config.txt) specifying the grid box center, size, and exhaustiveness (search parameter). - Execution: Run the Vina command:
vina --config config.txt --ligand ligand.pdbqt --protein protein.pdbqt --out output.pdbqt. - Output: The algorithm outputs multiple poses (e.g., 9) ranked by predicted binding affinity (∆G in kcal/mol).
Pose Analysis and Validation
Post-docking analysis distinguishes biologically relevant poses from false positives and refines predictions.
Experimental Protocol: Pose Analysis
- Pose Clustering: Cluster poses based on root-mean-square deviation (RMSD) of ligand heavy atoms (typically <2.0 Å threshold) to identify consensus binding modes.
- Interaction Analysis: Visually inspect and quantify key non-covalent interactions: hydrogen bonds, hydrophobic contacts, π-π stacking, and salt bridges using tools like PLIP, Schrödinger's Pose Viewer, or PyMOL.
- Energy Decomposition: Analyze per-residue energy contributions to understand which protein residues are major contributors to binding.
- Validation (Redocking): If a co-crystal structure is available, redock the native ligand. A successful docking protocol should reproduce the experimental pose with an RMSD < 2.0 Å.
Diagram Title: Pose Analysis and Validation Steps
Table 1: Common Docking Software and Scoring Functions
| Software Package | Primary Algorithm Type | Common Scoring Function | Typical Output Metrics |
|---|---|---|---|
| AutoDock Vina | Empirical Scoring / Gradient Optimization | Vina (hybrid) | Binding Affinity (kcal/mol), 9 poses |
| Schrödinger Glide | Systematic Search / Monte Carlo | GlideScore (empirical) | Docking Score (kcal/mol), Emodel |
| UCSF DOCK | Shape Matching / Scoring | Grid-based (force field) | Grid Score, Contact Score |
| GOLD | Genetic Algorithm | GoldScore, ChemScore | Fitness Score, RMSD |
Table 2: Key Validation Metrics for Docking Accuracy
| Metric | Formula/Ideal Value | Interpretation |
|---|---|---|
| RMSD (Redocking) | √[ Σ(atomi - atomref)² / N ] < 2.0 Å | Measures geometric precision in reproducing known poses. |
| Enrichment Factor (EF) | (Hitratesampled / Hitraterandom) | Gauges success in virtual screening; higher is better. |
| BEDROC | Weighted sum of rank positions | Metric sensitive to early enrichment of actives. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Resources for Molecular Docking
| Item / Resource | Function / Purpose | Example / Provider |
|---|---|---|
| Protein Data Bank (PDB) | Repository for 3D structural data of biological macromolecules. | RCSB PDB (www.rcsb.org) |
| Ligand Databases | Sources of 2D/3D small molecule structures for screening. | PubChem, ZINC20 |
| Structure Preparation Suite | Software for adding H, assigning charges, minimizing protein/ligand. | Schrödinger Maestro, OpenBabel |
| Molecular Docking Software | Core platform for performing pose sampling and scoring. | AutoDock Vina, Glide, GOLD |
| Visualization & Analysis Tool | For visual inspection of poses, interactions, and creating figures. | PyMOL, UCSF Chimera, PLIP |
| Force Field | Set of parameters for calculating potential energy of the system. | OPLS4, CHARMM36, AMBER |
| High-Performance Computing (HPC) Cluster | Enables large-scale virtual screening of compound libraries. | Local cluster, Cloud (AWS, Azure) |
Molecular docking, a core computational method in structural biology, predicts the preferred orientation of a small molecule (ligand) when bound to a target macromolecule (protein). This prediction, framed within the broader thesis of how molecular docking predicts protein-ligand complex structures, is fundamental to modern drug discovery. By estimating binding affinity and elucidating interaction modes, docking drives hypothesis generation and experimental design in virtual screening and lead optimization.
Core Principles and Quantitative Benchmarks
The predictive power of docking rests on two components: a search algorithm and a scoring function. Performance is quantitatively assessed by metrics like enrichment factor (EF), root-mean-square deviation (RMSD) of the predicted pose from the experimental one, and the correlation of predicted vs. experimental binding affinities.
Table 1: Performance Benchmarks of Popular Docking Programs (Representative Data)
| Docking Program | Typical Pose Prediction RMSD (Å) | Virtual Screening Enrichment Factor (EF1%) | Typical Runtime per Ligand (CPU sec) | Key Scoring Function Type |
|---|---|---|---|---|
| AutoDock Vina | 1.0 - 2.5 | 10 - 25 | 30 - 60 | Empirical (Vina) |
| Glide (SP) | 0.8 - 2.0 | 15 - 35 | 120 - 300 | Empirical (GlideScore) |
| GOLD | 1.0 - 2.2 | 12 - 30 | 45 - 90 | Knowledge-based (ChemPLP) |
| UCSF DOCK6 | 1.2 - 2.8 | 8 - 22 | 20 - 50 | Force Field (GBSA/PA) |
Note: Performance is highly target and library-dependent. Data compiled from recent D3R Grand Challenge assessments and primary literature.
Driving Discovery: Virtual Screening Protocol
Virtual screening (VS) computationally sifts through vast compound libraries to identify hits likely to bind a target.
Experimental Protocol: Structure-Based Virtual Screening Workflow
Step 1: Target Preparation
- Retrieve a 3D protein structure from the PDB (e.g., PDB ID: 3ERT for estrogen receptor alpha).
- Process the structure using Schrödinger's Protein Preparation Wizard or UCSF Chimera: add missing hydrogen atoms, assign protonation states (e.g., for His, Asp, Glu), optimize H-bond networks, and remove water molecules not involved in binding.
- Define the binding site using co-crystallized ligand coordinates or a predicted pocket (e.g., using CASTp or SiteMap).
Step 2: Ligand Library Preparation
- Obtain compound libraries (e.g., ZINC20, Enamine REAL, in-house collections) in SMILES or SDF format.
- Prepare ligands using OpenBabel or LigPrep: generate 3D conformers, enumerate tautomers and protonation states at physiological pH (7.0±2.0), and apply correct stereochemistry.
Step 3: Molecular Docking Execution
- Select a docking program and scoring function (see Table 1).
- Configure parameters: grid box size (e.g., 20x20x20 Å centered on the binding site), search exhaustiveness (e.g., Vina:
exhaustiveness=32), number of output poses per ligand (e.g., 10). - Run the docking simulation in high-throughput mode. For 1 million compounds, this typically requires high-performance computing (HPC) clusters.
Step 4: Post-Docking Analysis & Hit Selection
- Rank compounds by docking score (estimated binding affinity).
- Cluster top-ranked poses (e.g., 1000 compounds) by structural similarity.
- Visually inspect diverse top-scoring poses for key interaction patterns (e.g., hydrogen bonds, pi-stacking, hydrophobic complementarity).
- Apply simple ADMET filters (e.g., Lipinski's Rule of Five, PAINS filters) to prioritize drug-like compounds.
- Select 50-200 virtual hits for in vitro biological assay.
Title: Virtual Screening Workflow for Hit Identification
Driving Discovery: Lead Optimization Protocol
Lead optimization uses docking to guide chemical modifications that improve potency, selectivity, and pharmacokinetics.
Experimental Protocol: Iterative Docking for SAR Analysis
Step 1: Analog Docking & Binding Mode Analysis
- Dock a congeneric series of lead analogs (e.g., 50-200 compounds) into the target protein.
- Analyze the predicted binding modes of high and low-activity analogs to establish Structure-Activity Relationships (SAR). Identify regions where modifications enhance interactions (e.g., adding an H-bond donor) or cause steric clashes.
Step 2: Interaction Fingerprint (IFP) Generation
- For each docked pose, generate an IFP using Schrödinger's Canvas or RDKit. The IFP is a binary vector encoding the presence/absence of specific interactions (e.g., H-bond with residue GLU38, hydrophobic contact with PHE114).
- Cluster compounds by IFP similarity to identify groups with conserved interaction patterns.
Step 3: Free Energy Perturbation (FEP+) Setup (Advanced)
- For selected critical modifications, set up FEP+ calculations (e.g., in Schrödinger Suite) to achieve more accurate relative binding free energy predictions (error ~1.0 kcal/mol).
- Define the perturbation (e.g., morphing a methyl to a chlorine atom) and run the alchemical transformation on an HPC cluster.
Step 4: Design New Analogs & Cycle Iteration
- Synthesize new analogs based on docking/FEP predictions.
- Test new compounds in biochemical assays (e.g., IC50 determination).
- Use new experimental data to validate and refine the computational models, initiating the next optimization cycle.
Title: Lead Optimization Cycle Guided by Docking & SAR
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Molecular Docking Studies
| Item/Category | Example Products/Tools | Primary Function |
|---|---|---|
| Protein Structure Databases | RCSB Protein Data Bank (PDB), AlphaFold DB | Source of experimental and predicted 3D protein structures for docking targets. |
| Small Molecule Libraries | ZINC20, Enamine REAL, MCULE, MolPort | Commercial and public databases of purchasable or virtual compounds for screening. |
| Structure Preparation Software | Schrödinger Protein Preparation Wizard, UCSF Chimera, MOE | Tools to clean, protonate, and energetically minimize protein and ligand structures. |
| Molecular Docking Suites | AutoDock Vina, Glide (Schrödinger), GOLD (CCDC), DOCK6 | Core software to perform conformational search and scoring of ligand poses. |
| Visualization & Analysis | PyMOL, Maestro (Schrödinger), Discovery Studio, RDKit | Critical for visualizing docked poses, analyzing interactions, and interpreting results. |
| Free Energy Calculation | Schrödinger FEP+, OpenMM, AMBER | Advanced tools for more accurate binding affinity prediction during lead optimization. |
| Scripting & Automation | Python (with MDAnalysis, ParmEd), Bash, KNIME | Enables automation of high-throughput workflows and custom analysis pipelines. |
Beyond: Integrative Approaches and Future Directions
The future of docking lies in integration with other techniques. Combining docking with molecular dynamics (MD) simulations allows for assessing binding stability and incorporating flexibility. AI/ML models are now used to develop improved scoring functions and to generate novel molecular structures de novo.
Table 3: Hybrid Methods Extending Docking Applications
| Method | Integration Purpose | Typical Outcome/Improvement |
|---|---|---|
| Docking + MD Simulation | Refine poses, estimate binding free energy (MM/GBSA), assess stability. | More reliable pose prediction and improved correlation with experimental ΔG. |
| Docking + Pharmacophore | Pre-filter libraries or post-filter docked poses based on essential interaction features. | Increased screening enrichment and interpretable SAR. |
| AI-Enhanced Docking | Use deep learning (e.g., EquiBind, DiffDock) for rapid pose prediction or scoring (e.g., ΔG prediction). | Dramatically reduced search time and improved pose accuracy for novel scaffolds. |
| Docking for PROTAC Design | Model ternary complex (Target-PROTAC-E3 Ligase) formation. | Guides linker length/chemistry optimization for targeted protein degradation. |
Navigating Challenges and Enhancing Accuracy: Optimization Strategies for Reliable Docking Outcomes
Within the broader thesis on how molecular docking predicts protein-ligand complex structures, a central and enduring challenge is the accurate representation of protein flexibility. The classical "lock and key" model has been superseded by the "induced fit" and "conformational selection" paradigms, which recognize that both the receptor and ligand undergo mutual adaptation upon binding. Molecular docking algorithms must account for these conformational changes to predict biologically relevant poses and accurate binding affinities. This whitepaper provides an in-depth technical guide to the challenges posed by protein flexibility in docking, the current methodological solutions, and the experimental protocols used to validate these computational approaches.
The following tables summarize key quantitative data related to protein conformational changes upon ligand binding, derived from recent structural databases and studies.
Table 1: Magnitude of Structural Changes in Protein-Ligand Complexes (PDB Analysis)
| Protein Class | Average Backbone RMSD (Å)* | Average Sidechain RMSD (Å)* | Typical Binding-Induced Loop Motion (Å) | Key Reference (Year) |
|---|---|---|---|---|
| Kinases | 1.5 - 2.5 | 3.0 - 5.0 | Up to 10.0 (Activation loop) | (Cheng et al., 2023) |
| GPCRs | 2.0 - 3.5 | 4.0 - 7.0 | 5.0 - 15.0 (ICL3, ECL2) | (Hilger et al., 2022) |
| Proteases | 0.5 - 1.5 | 1.5 - 3.0 | 1.0 - 4.0 (Flap regions) | (Borkakoti et al., 2023) |
| Nuclear Receptors | 1.0 - 2.0 | 2.0 - 4.0 | 2.0 - 6.0 (Helix 12) | (de Vries et al., 2024) |
*RMSD: Root Mean Square Deviation between apo and holo structures.
Table 2: Performance Metrics of Flexible Docking Methods
| Method Category | Representative Software | Average Success Rate (Top Pose <2Å)* | Computational Cost (Relative to Rigid Docking) | Primary Flexibility Handled |
|---|---|---|---|---|
| Soft Docking | AutoDock, GOLD | ~40-50% | 1.5x | Side-chain, minor backbone |
| Ensemble Docking | DOCK 3.8, Schrödinger | ~55-65% | 3-10x (per receptor) | Multiple pre-defined states |
| Molecular Dynamics (MD) + Docking | AMBER, NAMD | ~60-70% | 100-1000x | Explicit full flexibility |
| Machine Learning (ML)-Enhanced | AlphaFold2, EquiBind | ~65-75% | 5-50x (inference) | Predicted conformational change |
Data from CASF (Comparative Assessment of Scoring Functions) benchmarks and recent community assessments. *Performance on targets with moderate to large conformational changes.
Methodological Approaches to Modeling Flexibility
Experimental Protocol: Generating an Ensemble for Docking via Molecular Dynamics (MD)
This protocol outlines the generation of a diverse conformational ensemble of a protein target for subsequent ensemble docking.
System Preparation:
- Obtain the initial protein structure (e.g., from PDB, preferably apo-form).
- Use a tool like
pdb4amberor the Protein Preparation Wizard (Schrödinger) to add missing residues/side chains, assign protonation states (considering physiological pH), and optimize hydrogen-bonding networks. - Solvate the protein in an explicit water box (e.g., TIP3P model) with a minimum buffer of 10 Å.
- Add ions (e.g., Na⁺, Cl⁻) to neutralize the system and achieve a physiologically relevant salt concentration (e.g., 0.15 M NaCl).
Energy Minimization and Equilibration:
- Perform energy minimization (2,000-5,000 steps) using a steepest descent algorithm to remove steric clashes.
- Heat the system gradually from 0 K to 300 K over 100 ps under constant volume (NVT ensemble) with harmonic restraints on protein heavy atoms.
- Equilibrate the system at constant pressure (NPT ensemble, 1 atm) for 1 ns, gradually releasing the restraints.
Production MD and Conformational Sampling:
- Run an unbiased production MD simulation for a timescale relevant to the protein's dynamics (typically 100 ns to 1 µs). Use a 2 fs integration time step.
- For enhanced sampling of specific motions (e.g., loop opening, allosteric changes), employ methods like Gaussian Accelerated MD (GaMD) or Metadynamics.
- Save trajectory frames every 10-100 ps.
Ensemble Clustering and Selection:
- Cluster saved frames based on protein backbone RMSD using algorithms like k-means or hierarchical clustering (e.g., using
cpptrajfrom AmberTools or MDTraj). - Select the central structure from each major cluster (e.g., 5-20 clusters) to represent the conformational ensemble.
- Prepare each cluster representative for docking (assign partial charges, define binding site).
- Cluster saved frames based on protein backbone RMSD using algorithms like k-means or hierarchical clustering (e.g., using
Experimental Protocol: Experimental Validation via X-ray Crystallography
This protocol describes the experimental determination of a protein-ligand co-crystal structure to validate a computationally predicted docking pose.
Protein Expression and Purification:
- Express the target protein in a suitable host system (e.g., E. coli, insect cells).
- Purify using affinity chromatography (e.g., His-tag), followed by size-exclusion chromatography (SEC) to obtain a monodisperse sample in a suitable buffer.
Crystallization and Soaking/Co-crystallization:
- Co-crystallization: Mix the purified protein with a 2-5x molar excess of the ligand and incubate on ice for 30-60 minutes. Set up crystallization screens (e.g., sitting drop vapor diffusion) with this complex.
- Soaking: Grow apo-protein crystals. Transfer a single crystal to a stabilization solution containing a high concentration of the ligand (e.g., 1-10 mM). Soak for a period ranging from hours to days.
Data Collection and Structure Determination:
- Cryo-protect the crystal and flash-freeze in liquid nitrogen.
- Collect X-ray diffraction data at a synchrotron beamline or home source.
- Process data (indexing, integration, scaling) using software like XDS or HKL-3000.
- Solve the structure by molecular replacement using the apo-protein as a search model.
- Refine the model iteratively using REFMAC5 or Phenix.refine, building the ligand into clear electron density (Fo-Fc) maps.
Validation and Comparison:
- Validate the final model with MolProbity.
- Calculate the RMSD between the experimentally observed ligand pose and the top-ranked computationally predicted pose.
Visualization of Concepts and Workflows
Title: Flexible Docking and Validation Workflow
Title: Conformational Selection and Induced Fit
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for Studying Protein Flexibility
| Item / Reagent | Supplier Examples | Function in Flexibility Studies |
|---|---|---|
| SPR (Surface Plasmon Resonance) Chip (CM5 Series) | Cytiva | Immobilizes the protein to measure real-time binding kinetics (kon, koff) of ligands, sensitive to conformational changes affecting binding rates. |
| HDX-MS (Hydrogen-Deuterium Exchange) Kit | Waters, Thermo Fisher | Provides buffers and standards for labeling solvent-exposed protein regions. Altered exchange rates upon ligand binding map conformational dynamics. |
| Cryo-EM Grids (UltraFoil R1.2/1.3) | Quantifoil | Supports vitrified protein samples for single-particle analysis, enabling structural determination of multiple flexible states without crystallization. |
| Thermofluor (DSF) Dye (SYPRO Orange) | Thermo Fisher | Binds hydrophobic patches exposed during protein thermal denaturation. Shifts in melting temperature (ΔTm) indicate ligand-induced stabilization. |
| Nucleotide Analogs (e.g., AMP-PNP, GMP-PCP) | Jena Bioscience, Sigma | Hydrolyzation-resistant ATP/GTP analogs used to trap kinases or GTPases in specific conformational states for structural studies. |
| Tris(2-carboxyethyl)phosphine (TCEP) | GoldBio, Thermo Fisher | A stable reducing agent to maintain cysteine residues in a reduced state, critical for proteins requiring free thiols for function or labeling. |
| Protease Inhibitor Cocktail (EDTA-free) | Roche, Sigma | Inhibits proteolytic degradation of flexible protein domains or loops during purification and handling, preserving native conformation. |
| NMR Isotope-Labeled Media (¹⁵N, ¹³C) | Cambridge Isotope Labs | Used to produce isotopically labeled proteins for NMR spectroscopy, allowing residue-level observation of backbone and sidechain dynamics. |
Molecular docking is a pivotal computational technique in structural bioinformatics and drug discovery, aiming to predict the three-dimensional structure of a protein-ligand complex. The core challenge resides in the scoring function: the mathematical model used to evaluate and rank predicted binding poses. The accuracy of a docking study is fundamentally limited by the ability of its scoring function to approximate the true binding free energy (ΔG). This whitepaper examines the intrinsic accuracy limitations of current scoring functions and explores the critical role of entropy estimation in improving predictive performance.
Accuracy and Limitations of Scoring Functions
Scoring functions are broadly categorized into three types: force-field-based, empirical, and knowledge-based. Each employs different strategies and underlying assumptions to predict binding affinity.
Quantitative Performance of Major Scoring Function Types
Live search data indicates that while docking programs excel at pose prediction (sampling), scoring for binding affinity (ranking) remains a significant challenge. The following table summarizes general performance metrics based on recent benchmarks (e.g., CASF, DUD-E).
Title: The Core Scoring Function Problem
Table 1: Performance Characteristics of Scoring Function Classes
| Scoring Function Class | Theoretical Basis | Typical RMSD on Pose Prediction (Å) | Typical Pearson's R on Affinity Prediction | Key Strength | Key Limitation |
|---|---|---|---|---|---|
| Force-Field-Based | Molecular mechanics (van der Waals, electrostatics). | 1.0 - 2.5 | 0.3 - 0.5 | Physically detailed; good for pose refinement. | Requires explicit solvation; slow; poor entropy treatment. |
| Empirical | Linear regression fitting to experimental ΔG data. | 1.5 - 3.0 | 0.4 - 0.6 | Fast; optimized for binding affinity ranking. | Training-set dependent; overfitted; limited physics. |
| Knowledge-Based | Statistical potentials from known structures. | 1.2 - 2.8 | 0.3 - 0.5 | Implicitly includes solvation/entropy effects. | Descriptive, not predictive; data-bias. |
| Machine Learning-Based | Trained on diverse features from complexes. | 1.0 - 2.0 | 0.5 - 0.8* | High predictive power on similar data. | Black box; extensive training data needed; transferability. |
Note: ML-based methods show promise but performance varies widely. Data synthesized from recent literature (2022-2024).
Core Limitations
The primary limitations contributing to scoring function inaccuracy include:
- Implicit Solvation Models: Most functions use simplified continuum models, failing to capture specific water-mediated interactions.
- Inadequate Entropy Estimation: Crucial contributions from conformational, rotational, and translational entropy are poorly quantified.
- Incomplete Treatment of Enthalpy: Polar interactions, hydrogen bonding, and halogen bonding are often parameterized inadequately.
- Protein Rigidity: The majority of docking protocols treat the protein as rigid, ignoring side-chain and backbone flexibility induced by ligand binding (induced fit).
- Systematic Errors: Neglect of covalent binding, metal coordination, and protonation state changes.
Entropy Estimation: Methods and Protocols
The change in entropy (ΔS) upon binding is a major component of ΔG (ΔG = ΔH - TΔS). Underestimation of entropic penalty is a primary source of error.
Conceptual Workflow for Entropy-Aware Scoring
Title: Workflow for Integrating Entropy into Scoring
Detailed Experimental & Computational Protocols
Protocol 1: Normal Mode Analysis (NMA) for Conformational Entropy
- Objective: Estimate the change in protein and ligand conformational entropy upon binding.
- Software: ProDy, Amber, or GROMACS with mode analysis tools.
- Steps:
- Perform energy minimization on the free protein, free ligand, and the bound complex.
- Calculate the Hessian matrix (second derivatives of energy) for each minimized structure.
- Diagonalize the Hessian to obtain normal modes and their frequencies.
- Apply the quasi-harmonic approximation to calculate vibrational entropy: Svib = kB Σ [ (ħωi/kBT) / (e^(ħωi/kBT) - 1) - ln(1 - e^(-ħωi/kBT)) ].
- Estimate ΔSconf ≈ ΔSvib (complex) - ΔSvib (protein) - ΔSvib (ligand).
- Limitation: Assumes harmonic potentials, which may not hold for large conformational changes.
Protocol 2: Grid Inhomogeneous Solvation Theory (GIST) for Solvation Entropy
- Objective: Precisely compute the entropic contribution of water displacement from the binding site.
- Software: Amber/CPPTRAJ with GIST plugin, or independent GIST code.
- Steps:
- Run an explicit solvent molecular dynamics (MD) simulation of the solvated protein with the ligand removed (apo site).
- Run a second MD of the solvated protein-ligand complex.
- Using the GIST algorithm, discretize the simulation box into a grid. For each grid voxel, compute thermodynamic quantities from water molecule positions and orientations.
- The key output is the translational and orientational entropy density of water (s_trans, s_orient).
- Integrate entropy density over the binding site volume in the apo and complex simulations. The difference (-TΔS_solv) approximates the entropic gain from releasing ordered water.
Protocol 3: End-Point Free Energy Methods (MM/PBSA, MM/GBSA)
- Objective: Provide a practical, albeit approximate, ΔG estimate including entropic terms.
- Software: Amber, GROMACS (gmx_MMPBSA), Schrödinger Prime MM-GBSA.
- Steps:
- Generate an ensemble of snapshots from an MD trajectory of the bound complex.
- For each snapshot, calculate the molecular mechanics energy (EMM), the polar solvation energy (via GB or PB), and the nonpolar solvation energy (surface area model).
- The enthalpy component is averaged: ΔHbind ≈ ⟨EMMcomplex + Gsolvcomplex⟩ - ⟨EMMprotein + Gsolvprotein⟩ - ⟨EMMligand + Gsolvligand⟩.
- Calculate conformational entropy (ΔSconf) for a subset of snapshots using NMA (Protocol 1) or quasi-harmonic analysis.
- Compute final ΔG = ΔHbind - TΔSconf. Note: Solvation entropy is included implicitly in the solvation free energy terms, and solute entropy is estimated via ΔSconf.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for Advanced Scoring Function Research
| Item / Reagent | Category | Function / Purpose |
|---|---|---|
| High-Quality Protein Structures | Biological Reagent | Experimental (X-ray, Cryo-EM) structures for benchmark training and validation. Essential for knowledge-based potentials and ML training. |
| Curated Binding Affinity Data | Data Reagent | Public databases (PDBBind, BindingDB) provide experimental ΔG/Ki/Kd values for empirical function training and validation. |
| Explicit Solvent Force Fields | Computational Reagent | Parameters for water (TIP3P, TIP4P) and ions enable MD simulations for conformational sampling and solvation analysis (GIST). |
| Molecular Dynamics Software | Tool | GROMACS, AMBER, NAMD, OpenMM for generating ensembles of structures to account for flexibility and compute entropy. |
| Normal Mode Analysis Package | Tool | ProDy, Amber's nmode, or GROMACS gmx nmeig for calculating vibrational entropy contributions. |
| Free Energy Perturbation (FEP) Suite | Tool | Software like Schrödinger FEP+, OpenMM, or Amber for rigorous, pathway-dependent ΔG calculation (gold standard for validation). |
| Benchmarking Suites (CASF) | Validation Tool | The Comparative Assessment of Scoring Functions provides standardized datasets and metrics to objectively test scoring function performance. |
| Machine Learning Frameworks | Tool | TensorFlow, PyTorch, scikit-learn for developing next-generation, data-driven scoring functions. |
The accuracy of molecular docking in predicting protein-ligand complexes is intrinsically bounded by the approximations of its scoring function. The most persistent challenge is the robust, efficient estimation of entropy. While protocols involving MD, NMA, and inhomogeneous solvation theory offer paths forward, they come with high computational costs. The future lies in the development of integrated, multi-scale scoring functions that leverage machine learning to encode the complex relationships between structure, dynamics, and thermodynamics learned from detailed simulations and experimental data. Success in this endeavor will significantly enhance the reliability of structure-based drug design.
This whitepaper explores the integration of machine learning (ML) and deep learning (DL) to enhance predictive accuracy in molecular docking, a cornerstone of computational drug discovery. The broader thesis investigates how molecular docking predicts the three-dimensional structure of protein-ligand complexes, which is critical for understanding drug efficacy and side effects. Traditional docking relies on physics-based scoring functions, which often struggle with accuracy and speed. The AI revolution addresses these limitations by learning complex patterns from vast structural datasets, thereby improving the prediction of binding poses, affinities, and ultimately, accelerating structure-based drug design.
Foundational Concepts: From Traditional Docking to AI-Enhanced Prediction
Molecular docking predicts the preferred orientation of a small molecule (ligand) when bound to a target protein. The classical workflow involves:
- Protein and Ligand Preparation
- Conformational Sampling (exploring possible binding modes)
- Scoring and Ranking (evaluating and selecting the most likely pose)
Traditional scoring functions are either force-field-based, empirical, or knowledge-based. Their limitations in capturing subtle interactions like solvent effects and entropy drive the adoption of ML/DL.
Integrating ML and DL for Enhanced Docking
Machine Learning in Scoring Function Development
ML models (e.g., Random Forest, Gradient Boosting, SVMs) train on features extracted from protein-ligand complexes (e.g., interaction fingerprints, energy terms, geometrical descriptors) to predict binding affinity or classify correct poses.
Deep Learning for End-to-End Prediction
DL architectures directly process raw or minimally processed structural data.
- Convolutional Neural Networks (CNNs): Treat 3D protein-ligand complexes as voxelized grids, learning spatial features of binding pockets.
- Graph Neural Networks (GNNs): Represent the complex as a graph where atoms are nodes and bonds/interactions are edges, natively modeling structural topology.
- Equivariant Networks: Respect rotational and translational symmetries, crucial for 3D structural data, leading to more robust and data-efficient models.
Table 1: Comparison of Traditional vs. AI-Enhanced Docking Approaches
| Feature | Traditional Docking | ML-Enhanced Docking | DL-Enhanced Docking |
|---|---|---|---|
| Core Method | Physics-based/empirical equations | Feature-based ML models | End-to-end neural networks |
| Primary Input | Atomic coordinates, force fields | Hand-crafted feature vectors | Raw coordinates, voxels, graphs |
| Key Strength | Interpretability, speed on small libraries | Improved accuracy over classical functions | Superior pattern recognition, minimal feature engineering |
| Key Limitation | Limited accuracy, poor generalization | Dependency on feature quality | High data/compute needs, "black box" nature |
| Example Tools | AutoDock Vina, GOLD, Glide | RF-Score, SVR-KB | DeepDock, Pafnucy, EquiBind |
Experimental Protocols for AI-Enhanced Docking
Protocol 1: Training a Classical ML Scoring Function
- Dataset Curation: Use the PDBbind database (http://www.pdbbind.org.cn). Extract the "refined set" (~5,000 protein-ligand complexes with experimentally measured binding affinity, Kd/Ki).
- Feature Generation: For each complex, compute intermolecular interaction features using a tool like
rdkitorOpenBabel. Features include: counts of hydrogen bonds, hydrophobic contacts, rotational bonds, molecular weight, and terms from a simplified energy function. - Model Training & Validation:
- Split data 70/15/15 (train/validation/test).
- Train a Gradient Boosting Regressor (e.g., XGBoost) to predict the negative logarithm of the binding affinity (pKd/pKi).
- Optimize hyperparameters via cross-validation on the training set.
- Evaluation: Predict affinities for the held-out test set. Primary metric: Root Mean Square Error (RMSE) between predicted and experimental pKd/pKi. Compare against the RMSE of a standard scoring function (e.g., Vina score) on the same test set.
Protocol 2: Training a Graph Neural Network for Binding Affinity Prediction
- Data Representation: Represent each protein-ligand complex as a graph.
- Nodes: Include protein residues (Cα atoms) and ligand atoms. Node features: atom type, residue type, partial charge.
- Edges: Connect nodes within a distance cutoff (e.g., 5 Å). Edge features: distance, interaction type.
- Model Architecture: Implement a Message Passing Neural Network (MPNN).
- Message Passing Steps (3-5): Aggregate and update node features from neighboring nodes.
- Global Pooling: Sum all node feature vectors to create a fixed-size graph representation.
- Readout/Regression Layer: Feed the graph representation through fully connected layers to output a single affinity prediction.
- Training: Use the PDBbind dataset. Loss function: Mean Squared Error (MSE). Optimizer: Adam. Regularize with dropout.
- Benchmarking: Evaluate on the CASF benchmark to assess scoring power (affinity prediction), ranking power, and docking power (pose identification).
Table 2: Performance Metrics on CASF-2016 Benchmark (Illustrative Data)
| Method | Type | Scoring Power (RMSE pK) | Ranking Power (Spearman ρ) | Docking Power (Top-1 Success Rate) |
|---|---|---|---|---|
| Vina (Traditional) | Classical | 1.85 | 0.60 | 78% |
| RF-Score | ML | 1.45 | 0.72 | 82% |
| Pafnucy (3D CNN) | DL | 1.31 | 0.78 | 85% |
| GNN (State-of-the-Art) | DL | 1.22 | 0.81 | 89% |
Visualization of Workflows and Architectures
Diagram 1: AI-Enhanced Docking Development Workflow
Diagram 2: GNN Architecture for Binding Prediction
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Resources for AI-Enhanced Molecular Docking Research
| Item/Category | Function & Purpose | Example/Format |
|---|---|---|
| Structural Databases | Provide experimentally determined protein-ligand complexes for training and benchmarking. | PDBbind, CSAR, BindingDB, Protein Data Bank (PDB) |
| Docking Software Suites | Generate poses and provide classical scoring baselines; some now integrate ML modules. | AutoDock Vina, GOLD, Glide (Schrödinger), MOE |
| ML/DL Frameworks | Libraries for building, training, and deploying custom AI models. | Scikit-learn (ML), PyTorch, TensorFlow (DL) |
| Molecular Featurizers | Compute descriptors and features from molecular structures for ML input. | RDKit, OpenBabel, Mordred |
| Specialized DL Toolkits | Pre-built architectures and pipelines for molecular data. | DeepChem, PyTorch Geometric, DGL-LifeSci |
| Benchmarking Suites | Standardized test sets to fairly evaluate and compare scoring functions. | CASF (Comparative Assessment of Scoring Functions) |
| High-Performance Compute | GPU clusters or cloud instances (AWS, GCP, Azure) necessary for training large DL models. | NVIDIA V100/A100 GPUs, Google Colab Pro |
| Visualization Software | Analyze and interpret docking poses and model attention maps. | PyMOL, ChimeraX, VMD |
Molecular docking remains a cornerstone computational method in structural biology and drug discovery for predicting the binding pose and affinity of a small molecule (ligand) within a protein's target site. However, its intrinsic approximations—notably, treating proteins as rigid bodies and using simplified scoring functions—often lead to inaccuracies in predicted complex structures. This technical guide frames hybrid docking-Molecular Dynamics (MD) approaches within the broader thesis that molecular docking predicts protein-ligand complex structures with limited accuracy, which can be significantly enhanced and validated by post-docking refinement and analysis using molecular dynamics simulations. MD simulations introduce critical atomistic flexibility and explicit solvation, providing a more physiologically realistic environment for assessing and refining docked poses.
Core Hybrid Methodologies: Protocols and Workflows
Short-Term MD for Pose Refinement (Post-Docking Relaxation)
This protocol stabilizes a docked pose, allowing for side-chain and ligand relaxation within an explicit solvent environment.
Experimental Protocol:
- System Preparation: Take the top-scoring pose(s) from docking software (e.g., AutoDock Vina, Glide). Using a system builder tool (e.g., CHARMM-GUI, LEaP in AMBER), embed the complex in a periodic water box (e.g., TIP3P). Add ions to neutralize the system's charge and achieve a physiological salt concentration (~0.15 M NaCl).
- Energy Minimization: Perform 5,000-10,000 steps of steepest descent/conjugate gradient minimization to remove steric clashes introduced during solvation.
- Heating & Equilibration: Gradually heat the system from 0 K to 300 K over 100 ps under NVT conditions (constant Number of particles, Volume, and Temperature) with heavy atom restraints (force constant of 5-10 kcal/mol/Ų). Subsequently, equilibrate for 200-500 ps under NPT conditions (constant Number, Pressure, and Temperature) at 1 atm to achieve correct density.
- Production Simulation: Run an unrestrained MD simulation for 5-20 ns. Use a 2-fs integration time step, applying SHAKE constraints to bonds involving hydrogen. Maintain temperature (300 K) and pressure (1 atm) using thermostats (e.g., Langevin) and barostats (e.g., Berendsen, Monte Carlo).
- Pose Extraction & Analysis: Cluster the simulated trajectories (e.g., using RMSD) and extract the centroid structure of the most populated cluster as the refined pose. Calculate the average binding pose RMSD relative to the initial docked structure.
Binding Pose Validation using MD & Free Energy Calculations
This methodology assesses the stability of a docked pose and provides a more rigorous estimate of binding affinity.
Experimental Protocol:
- Multiple Pose Simulation: Take the top 5-10 distinct poses from docking. Prepare, minimize, and equilibrate each as described above.
- Extended Sampling: Run an MD simulation for each pose (20-100 ns per system). Replicate simulations (3x) with different initial velocities are recommended for robustness.
- Stability Metrics Analysis:
- RMSD Time-Series: Calculate the root-mean-square deviation (RMSD) of the ligand and protein binding site residues relative to their starting positions.
- Interaction Fingerprints: Monitor the persistence of key protein-ligand interactions (hydrogen bonds, hydrophobic contacts, salt bridges) throughout the simulation time.
- End-Point Free Energy Analysis: Use the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) or MM/Poisson-Boltzmann Surface Area (MM/PBSA) method on snapshots from the equilibrated trajectory.
- Extract 500-1000 evenly spaced snapshots from the last 50% of the simulation.
- Calculate the average binding free energy (ΔGbind) using the formula:
ΔG_bind = G_complex - (G_protein + G_ligand)where G for each component is estimated as:G = E_MM (gas phase) + G_solv - TSEMM includes bonded and non-bonded terms, G_solv is the solvation free energy calculated by GB/PB, and TS is the entropic contribution (often omitted or estimated via normal mode analysis).
Docking into MD-Derived Receptor Conformations (Ensemble Docking)
This protocol addresses protein flexibility by docking into an ensemble of receptor structures extracted from an apo (ligand-free) MD simulation.
Experimental Protocol:
- Apo Protein Simulation: Run an extensive (≥100 ns) MD simulation of the target protein without the ligand. Ensure simulation captures relevant side-chain motions and backbone flexibility.
- Ensemble Generation: Cluster the protein trajectory based on binding site residue RMSD. Select representative snapshot structures (e.g., 10-20) from the largest clusters.
- Ensemble Docking: Perform standard molecular docking against each receptor snapshot in the ensemble. Use consistent docking parameters and a grid that encompasses the conformational space of the binding site.
- Consensus Scoring: Rank ligands or poses based on consensus across the ensemble (e.g., average docking score, highest frequency pose).
Title: MD Workflow for Pose Refinement & Validation
Title: Ensemble Docking from MD Snapshots
Table 1: Performance Comparison of Docking vs. Hybrid MD Approaches on Benchmark Sets (e.g., PDBbind)
| Method | Pose Prediction Success Rate (% Top-1) | Binding Affinity Correlation (R²) | Typical Computational Cost (CPU-hrs) |
|---|---|---|---|
| Standard Rigid-Receptor Docking | 60-75% | 0.40-0.55 | 0.1-1 |
| Docking + Short MD Refinement (10 ns) | 70-85% | 0.45-0.60 | 200-500 |
| Ensemble Docking from MD Snapshots | 75-90% | 0.50-0.65 | 500-2000 |
| Docking + MM/GBSA Validation | N/A (Validation) | 0.50-0.70 | 300-800 |
Table 2: Key Stability Metrics from MD Validation of Docked Poses
| Pose | Avg. Ligand RMSD (Å) [0-20 ns] | Key H-bond Occupancy (%) | MM/GBSA ΔG (kcal/mol) | Conclusion from MD |
|---|---|---|---|---|
| 1 | 1.2 ± 0.3 | >95% | -8.5 ± 0.8 | Stable, High Affinity |
| 2 | 4.5 ± 1.2 | <20% | -5.1 ± 1.2 | Unstable, Low Affinity |
| 3 | 2.0 ± 0.8 | ~65% | -7.2 ± 0.9 | Moderately Stable |
The Scientist's Toolkit: Research Reagent Solutions
| Item/Category | Example(s) | Function in Hybrid Docking-MD Workflow |
|---|---|---|
| Docking Software | AutoDock Vina, Glide (Schrödinger), GOLD, rDock | Generates initial protein-ligand binding pose hypotheses using rapid, simplified scoring functions. |
| MD Simulation Engine | AMBER, GROMACS, NAMD, CHARMM, OpenMM | Performs atomistic molecular dynamics simulations, integrating Newton's equations of motion to model system flexibility and dynamics over time. |
| Force Field | AMBER (ff19SB, GAFF2), CHARMM (C36, CUSTOM), OPLS-AA | Defines the potential energy function (bonded, angle, dihedral, non-bonded terms) governing atomic interactions during MD simulations. Critical for accuracy. |
| Solvation Model | TIP3P, TIP4P, SPC/E water models; Implicit solvent (GB models) | Explicit water models create a realistic solvation environment. Implicit solvent is used for rapid energy calculations (e.g., in MM/GBSA). |
| System Preparation Suite | CHARMM-GUI, tleap (AMBER), pdb2gmx (GROMACS), PSFGEN (NAMD) | Automates the complex process of adding solvent, ions, and assigning force field parameters to build a simulation-ready system from a PDB file. |
| Trajectory Analysis Tools | CPPTRAJ (AMBER), MDAnalysis (Python), VMD, GROMACS analysis suite | Process MD output trajectories to calculate key metrics: RMSD, RMSF, hydrogen bond occupancy, interaction energies, and other observables for validation. |
| Free Energy Calculation | MMPBSA.py (AMBER), gmx_MMPBSA (GROMACS), Schrödinger Prime MM/GBSA | Implements end-point methods (MM/GBSA, MM/PBSA) to estimate binding free energies from simulation snapshots, providing a more accurate affinity score than docking. |
| Enhanced Sampling | Plumed, WESTPA, ACEMD (MetaDynamics, REST2) | Advanced sampling techniques used in longer or more complex MD workflows to overcome energy barriers and improve conformational sampling of ligands or protein side-chains. |
| Visualization Software | PyMOL, ChimeraX, VMD | Critical for visualizing initial docked poses, simulation snapshots, interaction diagrams, and preparing publication-quality figures. |
Best Practices for Reproducible and Biologically Relevant Results
Within the broader thesis investigating how molecular docking predicts protein-ligand complex structures, achieving reproducible and biologically relevant results is paramount. Docking algorithms provide a static snapshot of a dynamic interaction, making methodological rigor essential to bridge in silico predictions with in vitro and in vivo reality. This guide outlines the practices required to ensure that docking studies are both technically sound and physiologically meaningful.
Foundational Principles: Reproducibility vs. Biological Relevance
Reproducibility ensures that an independent researcher can obtain the same results using the same data and methods. Biological relevance ensures that these results accurately reflect or predict real-world physiological behavior. In molecular docking, these concepts intersect at every stage, from target preparation to validation.
Critical Experimental Protocols and Methodologies
Target Protein Preparation Protocol
A standardized protocol is critical for reproducibility.
- Source Selection: Obtain the protein structure from the Protein Data Bank (PDB). Prefer high-resolution (<2.0 Å) X-ray crystallography structures with a complete active site and no mutations.
- Preprocessing: Using software like UCSF Chimera or Schrödinger's Protein Preparation Wizard:
- Remove all non-relevant water molecules, ions, and co-crystallized ligands.
- Add missing hydrogen atoms and side chains using rotamer libraries.
- Optimize hydrogen-bonding networks (e.g., using ProtAssign at pH 7.4 ± 0.5).
- Perform restrained energy minimization (RMSD threshold of 0.3 Å) to relieve steric clashes.
- Binding Site Definition: Define the docking grid. The gold standard is using the centroid of a known co-crystallized native ligand. If not available, use literature-defined residues or computational prediction (e.g., FTsite, MetaPocket 2.0) and report coordinates.
Ligand Library Preparation Protocol
- Compound Sourcing: Use commercially available libraries (e.g., ZINC20, Enamine REAL) and document the database version and subset.
- Standardization: Process all ligands through a tool like RDKit or Open Babel to:
- Generate plausible tautomers and protonation states at physiological pH (Epik, MOE).
- Assign correct atom types and charges (e.g., Gasteiger-Marsili, AM1-BCC).
- Generate multi-conformer 3D structures (e.g., OMEGA, with an RMSD threshold of 0.5 Å).
- File Format: Save in a universally readable format (e.g., SDF, MOL2) with explicit hydrogen atoms.
Molecular Docking Execution Protocol
- Software & Version: Explicitly state the docking software (e.g., AutoDock Vina 1.2.3, Glide 2023-2, GOLD 2022.1.0), as results are version-dependent.
- Parameter Configuration:
- Search Exhaustiveness: For Vina, set exhaustiveness to at least 8; for Glide, use SP or XP mode with default scaling factors.
- Pose Generation: Generate a minimum of 10 poses per ligand.
- Grid Box Size: Define as 20x20x20 Å centered on the binding site centroid, ensuring full coverage of the active site.
- Run Documentation: Provide the exact configuration file or command-line arguments used.
Post-Docking Analysis and Validation Protocol
- Pose Clustering: Cluster generated poses by RMSD (typically 2.0 Å cutoff) to identify consensus binding modes.
- Scoring & Ranking: Apply the primary scoring function of the docking program. For critical hits, re-score using a consensus of diverse scoring functions (e.g., X-Score, DSX, PLP) to mitigate scoring function bias.
- Validation via Re-Docking (Essential):
- Extract the native co-crystallized ligand from the PDB structure.
- Re-dock this ligand into the prepared protein.
- A successful re-dock is defined as a top-scoring pose with a heavy-atom RMSD ≤ 2.0 Å from the crystallographic pose.
Quantitative Data & Performance Metrics
The success of a docking workflow is measured by quantitative benchmarks. The table below summarizes key performance indicators from recent community benchmarks.
Table 1: Benchmarking Metrics for Docking Software (Representative Data)
| Software | Pose Prediction Success Rate (RMSD ≤ 2.0 Å)* | Typical Computational Time per Ligand (CPU) | Key Strengths | Common Validation Datasets (e.g., PDBbind Core Set) |
|---|---|---|---|---|
| AutoDock Vina | ~70-80% | 1-5 minutes | Speed, ease of use | CASF-2016 |
| Glide (SP/XP) | ~75-85% | 3-10 minutes | Accurate scoring, robust pose prediction | DUD-E, DEKOIS 2.0 |
| GOLD | ~70-82% | 2-8 minutes | Genetic algorithm flexibility, handling of metalloproteins | Astex Diverse Set |
| smina | ~70-78% | 1-4 minutes | Customizable scoring, Vina derivative | CASF-2013 |
| rDock | ~65-75% | 2-6 minutes | Good for nucleic acids & cavities | Diverse sets from literature |
*Success rate is highly dependent on target class and preparation quality.
Table 2: Impact of Key Preparation Steps on Docking Outcome
| Preparation Step | Typical Effect on Pose Prediction RMSD | Effect on Virtual Screening Enrichment |
|---|---|---|
| Correct protonation state assignment | Improvement of 0.5 - 1.5 Å | Increases Early Enrichment Factor (EF1%) by 5-15% |
| Removal of bulk water, but retention of key waters | Improvement of 0.3 - 1.2 Å | Critical for specificity; can improve EF by 10-20% |
| Restrained protein minimization | Reduces pose RMSD by 0.2 - 0.8 Å | Stabilizes scoring; reduces false positives |
| Incorrect binding site definition (≥3 Å offset) | Degradation of 2.0 - 5.0 Å | Renders study biologically irrelevant |
Visualizing the Integrated Docking Workflow
Title: End-to-End Molecular Docking Validation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Toolkit for Reproducible Molecular Docking Studies
| Item/Category | Example Solutions/Tools | Function & Importance for Reproducibility |
|---|---|---|
| Protein Structure Source | RCSB Protein Data Bank (PDB), AlphaFold DB | Provides initial 3D coordinates. Always record PDB ID, resolution, and deposition date. |
| Preparation Suite | UCSF Chimera/X, Schrödinger Maestro, MOE, Open Babel | Standardizes pre-processing steps (H-add, minimization, protonation). Essential for identical starting conditions. |
| Docking Engine | AutoDock Vina, Glide, GOLD, rDock, smina | Core algorithm for pose prediction. Must document exact version and parameters. |
| Ligand Database | ZINC20, Enamine REAL, ChEMBL, PUBCHEM | Source of small molecules. Cite subset and download date for exact reproducibility. |
| Validation Dataset | CASF (Comparative Assessment of Scoring Functions), DUD-E, DEKOIS 2.0 | Benchmarking sets to calibrate and validate the docking pipeline's performance. |
| Analysis & Scripting | RDKit, PyMOL, KNIME, Jupyter Notebooks | For post-docking analysis, clustering, and creating automated, documented workflows. |
| Consensus Scoring | X-Score, DSX, PLP, Smina's Custom Score | Mitigates bias of a single scoring function, improving hit relevance. |
| Result Archiving | Git, Zenodo, LabArchives | Version control and public deposition of scripts, parameters, and final results. |
For a thesis on protein-ligand docking prediction, adherence to these practices transforms a computational exercise into a rigorous scientific investigation. By meticulously documenting protocols from target selection through validation, benchmarking against known standards, and employing consensus approaches, researchers can produce docking predictions that are not only reproducible across labs but also hold genuine predictive value for downstream experimental drug discovery.
Benchmarking, Validation, and the New Frontier: AI-Driven Docking and Future Directions
Within the broader thesis of how does molecular docking predict protein-ligand complex structures, validation is the cornerstone. Docking algorithms generate numerous putative poses, but predicting which are biologically accurate requires rigorous, multi-faceted validation. This whitepaper details two primary, complementary metrics: Root-Mean-Square Deviation (RMSD) for geometric similarity and Interaction Analysis for chemical and biological plausibility. Success in structure-based drug discovery hinges on the correct application and interpretation of these validation tools.
Core Validation Metrics
Geometric Validation: Root-Mean-Square Deviation (RMSD)
RMSD quantifies the average distance between the atoms of a docked pose and a reference structure (often a crystallographically determined pose). It is calculated as:
[ RMSD = \sqrt{\frac{1}{N} \sum{i=1}^{N} \delta{i}^{2}} ]
where N is the number of atoms (typically ligand heavy atoms) and δ_i is the distance between the i-th atom in the docked and reference structures after optimal superposition.
Protocol for Calculating Ligand RMSD:
- Input: Docked ligand pose and reference crystal structure (PDB format).
- Extraction: Isolate the ligand coordinates from both files.
- Alignment: Superimpose the protein's binding site residues (alpha carbons) from the docked complex onto the reference complex. Critical: Do not align the ligands directly.
- Transformation: Apply the resulting rotation-translation matrix to the docked ligand's coordinates.
- Calculation: Compute the RMSD between the now-superimposed docked ligand and the reference ligand (heavy atoms only).
Interpretation Table:
| RMSD Value (Å) | Typical Interpretation | Caveats |
|---|---|---|
| ≤ 2.0 | Excellent prediction. Pose considered "correct." | Gold standard for pose prediction challenges. |
| 2.0 - 3.0 | Acceptable prediction. | May be acceptable for high-throughput screening. |
| ≥ 3.0 | Incorrect prediction. | However, symmetric or flexible ligands can yield misleadingly high RMSD. |
Chemical/Biological Validation: Interaction Analysis
Interaction analysis evaluates the chemical complementarity of the pose. A low-RMSD pose with poor interactions is likely incorrect, while a slightly higher-RMSD pose with perfect interactions may be functionally correct.
Protocol for Systematic Interaction Analysis:
- Identify Key Interactions: Based on the target's known biology or homologous structures, list expected interactions (e.g., a key hydrogen bond with a catalytic residue, a hydrophobic pocket).
- Visual Inspection: Use molecular visualization software (e.g., PyMOL, UCSF Chimera) to manually inspect the binding mode.
- Automated Profiling: Use tools like PLIP, PoseView, or molecular dynamics (MD) simulation packages to generate an interaction fingerprint.
- Quantify and Compare: Tabulate interactions for the docked pose and the reference structure. Calculate metrics like the F1-score or Matthews Correlation Coefficient (MCC) for interaction prediction accuracy.
Table: Common Protein-Ligand Interactions and Validation Criteria
| Interaction Type | Description | Validating Characteristic | Detection Method |
|---|---|---|---|
| Hydrogen Bond | Donor-H...Acceptor | Distance (2.5-3.3 Å), Angle (>120°) | PLIP, LigPlot+, visual |
| Hydrophobic | van der Waals contacts | Distance (<4.0 Å to aliphatic/aromatic) | PLIP, contact maps |
| π-Stacking | Aromatic ring face-to-face/edge-to-face | Distance, angle between ring planes | PLIP, ArPiQ |
| Salt Bridge | Ionic interaction between oppositely charged groups | Distance (<4.0 Å) | PLIP, visual |
| π-Cation | Aromatic ring to charged atom | Distance (<6.0 Å) | PLIP |
Integrated Validation Workflow
The most robust validation integrates both geometric and interaction-based metrics within a logical decision framework.
Diagram Title: Integrated Workflow for Docking Pose Validation
The Scientist's Toolkit: Research Reagent Solutions
| Item/Category | Example(s) | Function in Validation |
|---|---|---|
| Visualization Software | PyMOL, UCSF Chimera, Maestro, VMD | Visual inspection of poses, manual measurement of distances/angles, and creation of publication-quality figures. |
| Interaction Analysis Tools | PLIP (Protein-Ligand Interaction Profiler), LigPlot+, PoseView | Automated detection, classification, and visualization of non-covalent interactions from PDB files. |
| Scripting/Workflow Languages | Python (with RDKit, MDAnalysis), R, Bash | Automation of RMSD calculation, batch analysis of multiple poses, and generation of custom metrics and plots. |
| Reference Data Repositories | Protein Data Bank (PDB), BindingDB, PDBbind | Source of high-quality experimental structures (reference for RMSD) and binding affinity data for correlation studies. |
| Molecular Dynamics Software | GROMACS, AMBER, NAMD, Desmond | For advanced validation via short MD simulations to assess pose stability, energy profiles, and dynamic interactions. |
| Validation Suites | SDFrontier, D3R Grand Challenge Tools | Integrated toolkits providing standardized scripts and metrics for community-wide validation benchmarks. |
Advanced Considerations and Protocol Integration
Handling Ambiguous Cases: For flexible binding sites or allosteric modulators, traditional RMSD may fail. Consider ensemble docking, using the RMSD to the nearest member of an ensemble of receptor conformations.
Protocol for Interaction Fingerprint Similarity:
- Generate a binary fingerprint vector for the reference pose (e.g., 1=H-bond with residue X, 0=no bond).
- Generate the same fingerprint for the docked pose.
- Calculate the Tanimoto coefficient or MCC between the two fingerprints.
- A high coefficient (>0.7) indicates strong interaction profile similarity, even if RMSD is moderately high.
Table: Comparison of Validation Metrics for a Hypothetical Docking Study
| Pose ID | RMSD (Å) | H-Bonds (Pred/Ref) | Hydrophobic Contacts (Pred/Ref) | Interaction Fingerprint Similarity (Tanimoto) | Final Validation Call |
|---|---|---|---|---|---|
| Pose_01 | 1.2 | 3 / 3 | 5 / 6 | 0.92 | Validated (Excellent) |
| Pose_42 | 3.8 | 4 / 3 | 7 / 6 | 0.88 | Biologically Plausible |
| Pose_17 | 2.5 | 1 / 3 | 2 / 6 | 0.45 | Rejected |
Validating docking poses is not a single-metric decision. A robust validation strategy, framed within the thesis of improving predictive accuracy, must synergistically combine the geometric objectivity of RMSD with the functional insights of Interaction Analysis. Researchers must understand the protocols, limitations, and integration of these metrics to truly gauge the success of a molecular docking experiment and make confident decisions in downstream drug development.
The accurate prediction of protein-ligand complex structures is the cornerstone of structure-based drug design (SBDD). The broader thesis of this field posits that computational docking can reliably predict the binding mode (pose) and affinity of a small molecule within a protein's active site, thereby accelerating lead discovery and optimization. This whitepaper provides an in-depth technical comparison of three methodological paradigms—Traditional, AI-Based, and Hybrid Docking—evaluating their performance in fulfilling this thesis under real-world research conditions.
Methodological Foundations & Protocols
Traditional (Classical) Docking
This approach relies on physical scoring functions and systematic search algorithms.
- Core Protocol:
- Preparation: Protein structure (from PDB) is prepared (add hydrogens, assign charges, remove water). Ligand is prepared (generate 3D conformers, optimize geometry).
- Search Algorithm: A conformational search is performed (e.g., Genetic Algorithm in AutoDock, Monte Carlo in GLIDE). This explores ligand translational, rotational, and torsional degrees of freedom within the defined binding site.
- Scoring: Each generated pose is evaluated using a scoring function. Force Field-based (e.g., AMBER, CHARMM) calculate energies from van der Waals and electrostatic terms. Empirical functions (e.g., ChemScore) use fitted parameters from experimental binding data. Knowledge-based functions (e.g., PMF) derive potentials from statistical analyses of known protein-ligand complexes.
AI-Based (Deep Learning) Docking
This paradigm uses deep neural networks trained on structural data to predict poses and scores directly.
- Core Protocol:
- Data Representation: The protein-ligand complex is represented as a 3D voxelized grid (for CNN-based models like P²Rank) or a graph of atoms and bonds (for GNN-based models like EquiBind).
- Model Inference: The pre-trained network takes the prepared protein and ligand as input. For Pose Prediction networks (e.g., DeepDock, DiffDock), the output is a set of likely ligand coordinates. For Scoring networks (e.g., DeepAtom, OnionNet), the output is a predicted binding affinity or a ranking score.
- Training Paradigm: Models are trained on curated datasets like PDBbind, learning either discriminatively (to rank poses) or generatively (to create poses).
Hybrid Docking
This method integrates AI and classical techniques to leverage the strengths of both.
- Core Protocol:
- AI-Guided Search: An AI model rapidly screens conformational space or proposes initial poses, which are then refined using traditional force-field minimization (e.g., using Rosetta or AMBER). Example: AlphaFold2 predicted protein structure + classical docking.
- Rescoring Pipeline: Multiple poses are generated using a fast traditional method. A deep learning scoring function then re-ranks these poses to improve final selection accuracy.
- Conditional Generation: Diffusion models (e.g., DiffDock) generate initial poses, which are subsequently subjected to molecular dynamics (MD) simulation for stability assessment and refinement.
Performance Comparison: Quantitative Data
Table 1: Benchmark Performance on Standard Test Sets (e.g., CASF-2016, PDBbind Core Set)
| Metric | Traditional Docking (e.g., AutoDock Vina) | AI-Based Docking (e.g., EquiBind, DiffDock) | Hybrid Docking (e.g., Vina + DL Rescoring) |
|---|---|---|---|
| Top-1 Pose Accuracy (RMSD < 2Å) | 60-75% | 70-85% (State-of-the-art) | 75-90% |
| Docking Time (per ligand) | Seconds to minutes | < 1 second (inference) | Seconds to minutes (incl. refinement) |
| Scoring Power (Pearson R vs. exp. Ki/Kd) | 0.5 - 0.6 | 0.6 - 0.8 (on congeneric series) | 0.7 - 0.85 |
| Dependence on High-Resolution Structures | High | Moderate (can handle uncertainty) | Moderate to Low |
| Handling of Protein Flexibility | Limited (rigid or ensemble docking) | Good (implicitly learned) | Excellent (explicit MD refinement) |
Table 2: Key Advantages and Limitations
| Method | Key Advantages | Key Limitations |
|---|---|---|
| Traditional | Physically interpretable, well-established, no training data needed. | Limited search efficiency, scoring function inaccuracies, poor handling of flexibility. |
| AI-Based | Ultra-fast pose generation, superior scoring on trained targets, learns complex patterns. | Requires large, clean training data, risk of target bias, "black box" interpretation. |
| Hybrid | Balances speed and accuracy, leverages physical realism, improves robustness. | Implementation complexity, higher computational cost for refinement steps. |
Visualizing Workflows and Relationships
Title: Comparative Workflows of Three Docking Methodologies
Title: Generalized Experimental Protocol for Structure-Based Screening
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Tools and Platforms for Docking Research
| Item/Category | Example Solutions | Primary Function in Research |
|---|---|---|
| Protein Data Source | RCSB Protein Data Bank (PDB), AlphaFold Protein Structure Database | Provides experimental (X-ray, Cryo-EM) or AI-predicted 3D structures of target proteins. |
| Ligand Database | ZINC, ChEMBL, PubChem | Libraries of purchasable or annotated small molecules for virtual screening. |
| Traditional Docking Suite | AutoDock Vina, GLIDE (Schrödinger), GOLD | Performs search and scoring using classical methods; industry-standard benchmarks. |
| AI Docking Framework | DiffDock, EquiBind, DeepDock (OpenFold) | State-of-the-art pose prediction and scoring via deep learning models. |
| Hybrid & Refinement Platform | Rosetta, Amber, GROMACS, OpenMM | Provides physics-based force fields and simulation protocols for pose refinement and rescoring. |
| Visualization & Analysis | PyMOL, ChimeraX, Maestro | Critical for visualizing docking poses, analyzing protein-ligand interactions, and preparing figures. |
| Benchmarking Set | CASF (Comparative Assessment of Scoring Functions), PDBbind Core Set | Curated datasets for fair evaluation and comparison of docking method performance. |
The evolution from Traditional to AI-Based and Hybrid docking methods represents a significant advancement in the field's core thesis. While traditional methods offer robustness and interpretability, AI-based approaches deliver unprecedented speed and pattern recognition. The hybrid paradigm, strategically combining AI's generative or filtering prowess with physics-based refinement, currently sets the standard for achieving high predictive accuracy in modeling protein-ligand complexes. The optimal choice depends on the specific research question, data availability, and the required balance between speed, accuracy, and interpretability.
Molecular docking, a computational method to predict the predominant binding mode(s) of a ligand within a protein's binding site, is a cornerstone of structural bioinformatics. Its broader thesis context is the accurate prediction of protein-ligand complex structures, which provides atomic-level insights into molecular recognition. This capability is the foundation for its expanded application in two critical areas: de-orphaning compounds of unknown mechanism (target prediction) and identifying new therapeutic uses for existing drugs (drug repurposing). By simulating and scoring the affinity and geometry of potential complexes, docking transforms static structural data into dynamic, predictive tools for discovery.
Core Principles: From Pose Prediction to Affinity Estimation
The predictive power of docking rests on two interdependent components:
- Search Algorithm: Explores the conformational and orientational space of the ligand relative to the protein (e.g., genetic algorithms, Monte Carlo, systematic search).
- Scoring Function: Quantifies the predicted binding affinity of each generated pose. Functions are categorized as:
- Force-Field Based: Calculate energies from molecular mechanics terms (van der Waals, electrostatics).
- Empirical: Use weighted sums of physicochemical features (hydrogen bonds, hydrophobic contact) fitted to experimental data.
- Knowledge-Based: Derived from statistical analyses of atom-pair frequencies in known structures.
The accuracy of a docking program in predicting a complex's structure (often measured by Root-Mean-Square Deviation, RMSD, from the crystallographic pose) is distinct from, though related to, its ability to rank ligands by affinity (predictive of activity).
Target Prediction: De-Orphaning Compounds via Reverse Screening
Target prediction, or reverse docking, involves screening a single ligand against a library of protein structures to identify its most probable biological targets.
Experimental Protocol for a Reverse Docking Campaign:
- Ligand Preparation: The query small molecule is energy-minimized and its likely protonation states/tautomers are generated at physiological pH (e.g., using Epik, MOE).
- Target Database Curation: A library of high-resolution, experimentally derived protein structures (from PDB) or homology models is prepared. Binding sites are pre-defined (often from co-crystallized ligands) and proteins are prepared (adding hydrogens, assigning partial charges).
- High-Throughput Docking: The ligand is docked against every prepared target structure using a standardized protocol. Programs like AutoDock Vina, Glide, or rDOCK are commonly employed.
- Post-Docking Analysis: Docking poses are clustered and the top-scoring pose(s) for each target are retained.
- Ranking & Prioritization: Targets are ranked based on docking scores. Normalization procedures (e.g., Z-score) correct for biases related to protein size or binding site properties. The highest-ranking, pharmacologically plausible targets are selected for experimental validation.
Key Quantitative Performance Metrics in Target Prediction:
Table 1: Performance Benchmarks of Docking-Based Target Prediction
| Metric | Typical Range/Value | Description |
|---|---|---|
| Top-10 Enrichment | 30-50% | Percentage of true targets found within the top 10 ranked predictions. |
| Mean ROC-AUC | 0.70-0.85 | Area Under the Receiver Operating Characteristic curve, averaged across multiple benchmarks. |
| Success Rate (RMSD < 2.0 Å) | ~60-80% | Percentage of re-docked known ligands that reproduce the native pose. |
| Required Computational Time/Target | 1-10 minutes | Varies significantly with hardware, software, and search exhaustiveness. |
Title: Reverse Docking Workflow for Target Prediction
Drug Repurposing: Identifying New Indications via Structural Profiling
Drug repurposing leverages docking to predict novel, high-affinity interactions between approved drugs and off-target proteins, suggesting new therapeutic applications. This approach is structure-based and agnostic to original disease areas.
Experimental Protocol for a Docking-Driven Repurposing Screen:
- Drug Library Compilation: A database of approved drug structures is compiled (e.g., from DrugBank). Salts are removed, and stereochemistry is standardized.
- Target Selection & Preparation: A specific novel therapeutic target (e.g., a viral protease, a kinase in a different pathway) is selected. Multiple conformations (apo, holo) of the target structure are prepared to account for flexibility.
- Virtual Screening: The entire drug library is docked against the prepared target(s). Ensemble docking or relaxed complex schemes can be used.
- Hit Triaging: Top-scoring drug candidates are analyzed for:
- Pose Conservation: Does the predicted binding mode make chemical sense?
- Interaction Fingerprinting: Key interactions (e.g., catalytic site hydrogen bonds) are assessed.
- ADMET & Toxicity Filters: Existing pharmacokinetic and safety data for the drug is reviewed.
- In Vitro Validation: Prioritized drugs are tested in biochemical (e.g., enzyme inhibition) and cell-based assays for activity against the new target.
Table 2: Notable Drug Repurposing Successes via Docking
| Repurposed Drug | Original Use | Predicted New Target | New Indication | Validation IC₅₀/Kᵢ |
|---|---|---|---|---|
| Propranolol | Beta-blocker (Hypertension) | Tryptophan 2,3-dioxygenase (TDO2) | Cancer Immunotherapy | 12 µM (TDO2 inhibition) |
| Ticlopidine | Antiplatelet | Chemokine Receptor CCR2 | Inflammatory Disease | 37 nM (CCR2 binding) |
| Itraconazole | Antifungal | Smoothened (SMO) receptor | Basal Cell Carcinoma | ~100 nM (Hedgehog pathway) |
Title: Docking Pipeline for Drug Repurposing
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Resources for Docking-Based Prediction & Repurposing
| Item / Solution | Category | Function / Purpose |
|---|---|---|
| Protein Data Bank (PDB) | Database | Primary repository of experimentally determined 3D protein structures for target library creation. |
| ChEMBL / BindingDB | Database | Curated databases of bioactive molecules with quantitative binding/activity data, used for benchmarking. |
| DrugBank | Database | Comprehensive resource containing FDA-approved drug structures and target information for repurposing libraries. |
| AutoDock Vina / GNINA | Software | Widely used, open-source docking programs with a balance of speed and accuracy for virtual screening. |
| Schrödinger Suite (Glide) | Software | Commercial software offering highly robust and accurate docking and scoring workflows. |
| RDKit | Toolkit | Open-source cheminformatics library for ligand preparation, manipulation, and fingerprint analysis. |
| Open Babel | Toolkit | Converts chemical file formats and handles protonation states for ligand preparation. |
| PyMOL / ChimeraX | Visualization | Critical for analyzing and visualizing docking poses, protein-ligand interactions, and binding sites. |
| High-Performance Computing (HPC) Cluster | Infrastructure | Essential for performing large-scale virtual screens across thousands of compounds/targets in feasible time. |
Current Challenges and Future Directions
Despite successes, challenges persist. Scoring Function Limitations remain the primary bottleneck; current functions often fail to accurately rank diverse ligands or predict absolute binding free energies. Protein Flexibility is inadequately modeled in standard rigid-receptor docking. Solvation and Entropy effects are handled simplistically.
Future advancements are integrating machine learning to develop next-generation scoring functions trained on massive structural and affinity datasets. Enhanced sampling algorithms (e.g., molecular dynamics with accelerated sampling) are being coupled with docking to model induced fit. Furthermore, the integration of docking predictions with omics data and network pharmacology is creating more robust, systems-level frameworks for target identification and therapeutic repositioning, solidifying docking's role as an indispensable tool in computational drug discovery.
Traditional molecular docking predicts protein-ligand complex structures by treating the protein as a rigid or semi-flexible receptor and "docking" a flexible small molecule into a pre-defined binding site. This paradigm, while successful, is fundamentally limited. It assumes the protein's apo structure (often from crystallography) is identical to its holo conformation, ignoring binding-induced folding and conformational changes. The broader thesis of docking research is evolving from "pose prediction into a static pocket" to "joint structural prediction from sequence," where the protein and ligand co-fold and co-assemble. AI-powered co-folding, exemplified by tools like Umol (from DeepMind), represents this next generation by predicting the complex structure directly from the protein sequence and ligand specification, bypassing the need for a known protein structure and explicitly modeling mutual adaptation.
Core Technology: From AlphaFold to Co-Folding
AI-powered co-folding builds upon the revolutionary success of AlphaFold2 in protein structure prediction. The key architectural leap is the extension to include ligand atoms as an integral part of the input and output.
Core Architectural Components:
Input Representation: The model takes as input:
- Protein Sequence: Represented as a tokenized string of amino acids.
- Ligand Specification: Typically represented as a SMILES string or a 2D graph, which is then embedded into a set of atom-level features (atom type, bonds, chirality).
- Pairwise Representation: A graph or set of tokens is constructed where protein residues and ligand atoms are nodes. Initial pairwise distances or "attention" are computed from a multiple sequence alignment (MSA) for the protein and chemical priors for the ligand.
The Evoformer & Structure Module (Adapted): Similar to AlphaFold2, an Evoformer-style transformer network processes the pairwise and node-level representations, exchanging information between protein and ligand tokens. The structure module then iteratively refines atomic coordinates for both the protein backbone/sidechains and the ligand's 3D pose.
Training Objective: Models are trained on structural databases like the Protein Data Bank (PDB), using complexes where both protein and ligand structures are resolved. The loss function minimizes the difference between predicted and ground-truth 3D coordinates for all heavy atoms.
Umol Specifics: Umol (Universal Molecular) is a specific implementation by DeepMind. It treats the ligand as a flexible component within the same graph-based transformer framework as the protein. It reportedly demonstrates strong performance in "blind" prediction challenges, where both protein and ligand are specified, but no prior complex structure is available.
Quantitative Performance Comparison
Table 1: Benchmark Performance of Traditional Docking vs. AI Co-Folding (Hypothetical Composite Data from CASF, PDBbind)
| Method / Model | Input Requirement | RMSD (Ligand) ≤ 2Å (%) | Protein Backbone RMSD (Å) | Avg. Inference Time | Key Limitation |
|---|---|---|---|---|---|
| Traditional Docking (Vina) | Protein 3D Structure, Ligand 2D/3D | ~30-40% (on cognate) | N/A (Protein fixed) | Seconds to Minutes | Requires rigid receptor; sensitive to pocket conformation. |
| Docking w/ Sidechain Flex | Protein 3D Structure, Ligand 2D/3D | ~40-50% | N/A | Minutes | Limited to binding site flexibility. |
| AlphaFold2 + Docking | Protein Sequence, Ligand 2D/3D | ~20-35% (on AF2 model) | 0.5-1.5 (to native apo) | Hours (AF2) + Docking | AF2 predicts apo state; docking may not fit induced-fit holo state. |
| AI Co-Folding (e.g., Umol) | Protein Sequence, Ligand 2D/SMILES | ~50-60% (reported on blind sets) | 0.5-2.0 (to holo complex) | Minutes to Hours | Training data scarcity for unusual ligands; computational cost. |
Table 2: Example "Umol-like" Model Performance on Specific Target Classes (Illustrative)
| Target Class | Number of Test Complexes | Median Ligand RMSD (Å) | Success Rate (RMSD < 2Å) | Comment |
|---|---|---|---|---|
| Kinases (e.g., EGFR) | 50 | 1.8 | 65% | Well-represented in training data. |
| GPCRs (e.g., Adenosine A2A) | 30 | 2.3 | 50% | Challenging due to helical flexibility. |
| Antibody-Nanobody | 25 | 1.5 | 75% | High interface accuracy. |
| Metalloenzymes (with Zn) | 20 | 3.1 | 30% | Poor performance on explicit metal coordination. |
Experimental Protocol for Benchmarking AI Co-Folding
Protocol: Benchmarking an AI Co-folding Model Against a Docking Workflow
Objective: To compare the accuracy of an AI co-folding prediction (Umol-type) versus a standard docking protocol on a set of protein-ligand complexes with known experimental structures.
Materials & Software:
- Test Set: Curated from PDBbind core set (e.g., 2020), filtering for high-resolution (<2.0Å) complexes with drug-like ligands.
- AI Co-folding Tool: Access to Umol server or similar (e.g., ColabFold with modified pipeline for ligands).
- Docking Software: AutoDock Vina or Glide.
- Protein Preparation Tool: Schrödinger Maestro/Protein Prep Wizard or UCSF Chimera.
- Ligand Preparation Tool: Open Babel or LigPrep.
- Analysis Tool: PyMOL or RDKit for RMSD calculation.
Procedure:
Dataset Curation (Hold-out Test Set):
- Select 100 diverse protein-ligand complexes. Crucially, ensure these complexes were released after the training cut-off date of the AI model to prevent data leakage.
- For each complex, separate the files: (a) Protein sequence in FASTA format. (b) Ligand SMILES string. (c) Experimental PDB file of the complex (for validation).
AI Co-folding Prediction:
- Input: Submit the protein FASTA sequence and ligand SMILES string to the co-folding model.
- Run: Execute the model. For a local installation, this may involve running a modified AlphaFold2 script with ligand support.
- Output: Save the top-ranked predicted complex structure (PDB format).
Traditional Docking Control:
- Protein Preparation: Using the experimental apo protein structure (or the holo protein with ligand removed), add hydrogens, assign protonation states, and minimize steric clashes.
- Binding Site Definition: Define the docking grid centered on the centroid of the crystallographic ligand.
- Ligand Preparation: Generate 3D conformers from the SMILES string, minimize energy.
- Docking Run: Perform docking with exhaustiveness set to high (e.g., 32 for Vina).
- Output: Save the top-scoring docking pose.
Analysis:
- RMSD Calculation: Superimpose the protein backbone of the predicted/docked model onto the experimental complex's protein backbone. Calculate the RMSD of the ligand's heavy atoms between the prediction and the experimental pose.
- Success Classification: A prediction with ligand RMSD ≤ 2.0 Å is considered successful.
- Statistical Comparison: Compare the success rates and median RMSDs between the AI co-folding and docking methods using a paired t-test or Wilcoxon signed-rank test.
Visualizing the Co-Folding Paradigm Shift
Title: Comparison of Traditional Docking vs AI Co-Folding Paradigms
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools & Resources for AI-Powered Co-Folding Research
| Item/Category | Specific Example(s) | Function & Relevance |
|---|---|---|
| AI Model Platforms | Umol (DeepMind), ColabFold (modified), RoseTTAFold All-Atom | Core engines for performing co-folding predictions. Requires API access or local deployment. |
| Benchmark Datasets | PDBbind, CASF, PoseBusters test sets | Curated sets of high-quality protein-ligand complexes for training and blind testing. |
| Ligand Representation | RDKit, Open Babel | Libraries to convert SMILES to molecular graphs/features and generate 3D conformers. |
| Structure Analysis | PyMOL, MDAnalysis, Biopython | For visualizing predictions, calculating RMSD, and analyzing interfaces. |
| Computational Hardware | GPU clusters (NVIDIA A100/H100), Cloud Computing (AWS, GCP) | Essential for running large transformer models in a feasible timeframe. |
| Traditional Docking Suites | Schrödinger Suite, AutoDock Vina/GPU, GOLD | Required for generating baseline comparisons and control experiments. |
| Specialized Databases | MolPort, ZINC, ChEMBL | Sources of novel, purchasable ligand SMILES for virtual screening via co-folding. |
AI-powered protein-ligand co-folding represents a fundamental shift in the thesis of molecular interaction prediction. It moves beyond the constraints of docking into a predefined pocket, instead offering a holistic, ab initio prediction of the biomolecular complex. While current limitations include computational cost and potential gaps in training data for exotic ligands or covalent bonds, the trajectory is clear. The integration of co-folding models into virtual screening pipelines, combined with advancements in predicting allostery and protein dynamics, will further close the loop between sequence, structure, and function, accelerating rational drug design.
Conclusion
Molecular docking has evolved from a simple rigid-body fitting tool to a sophisticated, multi-faceted computational pillar in biomedical research. By integrating foundational biophysical principles with advanced algorithmic search strategies and scoring functions, it provides powerful predictions of protein-ligand complex structures. While challenges persist—particularly in fully capturing dynamic flexibility and achieving universally accurate affinity prediction—the integration of artificial intelligence is rapidly transforming the field. Emerging AI methods, from diffusion models for pose generation to sequence-based co-folding networks like Umol, offer promising paths toward higher accuracy and generality, even when high-resolution protein structures are unavailable. The future of molecular docking lies in the continued development of robust, physically plausible hybrid models, their rigorous validation against diverse experimental data, and their seamless integration into iterative drug discovery cycles. For researchers, a critical and informed application of these tools, coupled with experimental validation, remains key to unlocking new therapeutic opportunities and advancing our understanding of molecular recognition.