Block Relevance (BR) Analysis: A Practical Guide to Lipophilicity Assessment in Drug Discovery
Block Relevance (BR) analysis is a computational tool designed to deconvolute the balance of intermolecular interactions governing partitioning and retention phenomena, making the interpretation of lipophilicity more accessible for medicinal...
Block Relevance (BR) Analysis: A Practical Guide to Lipophilicity Assessment in Drug Discovery
Abstract
Block Relevance (BR) analysis is a computational tool designed to deconvolute the balance of intermolecular interactions governing partitioning and retention phenomena, making the interpretation of lipophilicity more accessible for medicinal chemists. This article explores the foundational principles of BR analysis, detailing its application in converting chromatographic data into reliable log P surrogates, characterizing challenging compounds like zwitterions, and optimizing methods for measuring permeability. By providing a framework for troubleshooting complex molecular properties and validating experimental techniques, BR analysis enhances the efficiency and reliability of drug candidate prioritization, supporting the development of compounds with improved drug-likeness and therapeutic potential.
Understanding Block Relevance Analysis: Deconvoluting Molecular Interactions for Medicinal Chemists
Block Relevance (BR) analysis is a computational tool that deconvolutes the balance of intermolecular interactions governing drug discovery-related phenomena described by QSPR/PLS models [1]. This methodology provides medicinal chemists with a practical framework for interpreting partitioning and retention phenomena, making the choice of methods for measuring lipophilicity and permeability safer and more efficient [2]. Within pharmaceutical sciences, accurate assessment of lipophilicity is crucial as it directly influences key drug properties including permeability, solubility, and metabolic stability [3].
The fundamental strength of BR analysis lies in its ability to graphically represent the relative contribution of different blocks of molecular descriptors in Partial Least Squares (PLS) models [3]. This factorization allows researchers to move beyond simple numerical results to understand the mechanistic forces driving molecular behavior, particularly in complex biological systems where multiple interaction forces operate simultaneously [1] [2].
Theoretical Framework and Methodology
Core Principles of BR Analysis
BR analysis operates on the principle that the intermolecular forces governing lipophilicity and permeability can be decomposed into distinct blocks of descriptors representing different physicochemical properties [3]. The methodology specifically addresses the challenge that lipophilicity indices from different measurement systems can only be interconverted when they express the same balance of intermolecular solute-system forces [2].
The analysis is performed using a specialized MATLAB implementation that processes QSPR/PLS models containing multiple molecular descriptors [1]. These descriptors are grouped into "blocks" representing different aspects of molecular interactions, such as hydrogen bond donation, hydrogen bond acceptance, molecular size, and hydrophobicity [3]. Through mathematical factorization, BR analysis quantifies how much each block contributes to the overall model, providing visual and numerical output that highlights the dominant forces in the system under investigation [2] [3].
Key Physicochemical Properties in Lipophilicity Assessment
Table 1: Core Physicochemical Properties Deconvoluted by BR Analysis
| Property Block | Molecular Interaction Represented | Role in Lipophilicity & Permeability |
|---|---|---|
| Hydrogen Bond Donor (HBD) | Ability to donate hydrogen bonds | Dominates Δlog Poct–tol; decreases membrane permeability [3] |
| Hydrogen Bond Acceptor (HBA) | Ability to accept hydrogen bonds | Influences solubility and permeation; part of Ro5 parameters [3] |
| Molecular Size/Shape | Steric bulk and molecular dimensions | Affects diffusion rates through membranes; part of Ro5 [3] |
| Hydrophobicity | Affinity for lipophilic environments | Governs partitioning into biological membranes [1] |
Experimental Protocols and Applications
Protocol 1: Determination of Reliable log Poct Surrogates
Objective: Identify the optimal chromatographic system that provides accurate surrogates for octanol/water partition coefficients (log Poct) [1].
Materials and Methods:
- Equipment: HPLC system with Supelcosil LC-ABZ or similar column [2]
- Software: MATLAB with BR analysis implementation [1]
- Samples: 36+ compounds with known physicochemical properties [2]
Procedure:
- Sample Preparation: Prepare stock solutions of test compounds in appropriate solvents at concentration of 1 mg/mL.
- Chromatographic Analysis: Inject samples and record retention times under isocratic conditions using mobile phases with varying pH and organic modifier composition.
- Data Conversion: Calculate chromatographic indices (log k) from retention times.
- BR Analysis Implementation:
- Input 82+ VolSurf+ descriptors for each compound into the BR analysis system [3]
- Group descriptors into six interpretable blocks representing different molecular interactions [3]
- Run PLS analysis to correlate chromatographic indices with reference log Poct values
- Generate BR visualization showing contribution of each descriptor block
- System Selection: Identify the chromatographic system whose BR profile most closely matches that of the reference octanol/water system [1]
Interpretation: A chromatographic system producing a BR analysis profile with similar block relevance weighting as the octanol/water system provides a reliable log Poct surrogate [1] [2].
Protocol 2: Assessment of Hydrogen Bond Acidity Using Δlog Poct–tol
Objective: Utilize Δlog Poct–tol (difference between log Poct and log Ptol) to determine hydrogen bond donor properties of drug candidates [3].
Materials and Methods:
- Reference Systems: Octanol/water and toluene/water partition systems [3]
- Dataset: 200+ compounds with experimental log Poct and log Ptol values [3]
- Software: BR analysis implementation with VolSurf+ descriptor calculation [3]
Procedure:
- Data Collection: Compile experimental log Poct and log Ptol values from literature sources, excluding data obtained using DMSO as cosolvent [3].
- Calculate Δlog Poct–tol: Compute difference values (log Poct – log Ptol) for each compound.
- Descriptor Calculation: Process all compounds through VolSurf+ software to generate 82 molecular descriptors [3].
- Dataset Curation: Remove molecules capable of forming intramolecular hydrogen bonds (IMHBs) using specialized in-house software [3].
- PLS Model Development: Correlate Δlog Poct–tol values with VolSurf+ descriptors using PLS regression.
- BR Analysis Implementation: Apply BR analysis to group descriptors into six interpretable blocks and determine their relative contributions to the model [3].
Interpretation: BR analysis demonstrates that hydrogen bond donor properties predominantly govern Δlog Poct–tol values, supporting its use for estimating HBD properties in drug discovery [3].
Workflow Visualization: BR Analysis in Lipophilicity Assessment
Diagram 1: BR Analysis Workflow for Lipophilicity Assessment
Protocol 3: Permeability Prediction Across Cell Types
Objective: Validate the universality of passive permeability measurements among different cell types and identify optimal PAMPA methods [1].
Materials and Methods:
- Cell Systems: Caco-2, MDCK, PAMPA variants [1]
- Instrumentation: Permeability assay equipment
- Software: BR analysis computational tool [1]
Procedure:
- Experimental Data Collection: Measure passive permeability for diverse compound sets across multiple cell systems and PAMPA variants.
- Descriptor Calculation: Compute molecular descriptors for all test compounds.
- Model Development: Construct PLS models correlating descriptors with permeability values for each system.
- BR Analysis Application: Perform BR analysis on each PLS model to deconvolute the balance of intermolecular forces governing permeability in each system.
- System Comparison: Compare BR profiles across different cell types and PAMPA variants to identify systems with equivalent intermolecular force balances [1].
Interpretation: PAMPA methods that produce BR profiles similar to cell-based systems provide equivalent information about the balance of intermolecular interactions governing permeability, enabling their use as reliable surrogates for more complex cellular assays [1].
Essential Research Reagents and Computational Tools
Table 2: Research Reagent Solutions for BR Analysis Applications
| Reagent/Resource | Function/Application | Specifications/Alternatives |
|---|---|---|
| Supelcosil LC-ABZ Column | Chromatographic determination of lipophilicity indices | Used with mobile phases of varying pH and organic modifier composition [2] |
| VolSurf+ Software | Calculation of molecular descriptors for PLS modeling | Generates 82+ descriptors covering various physicochemical properties [3] |
| BR Analysis MATLAB Implementation | Deconvolution of intermolecular interactions in PLS models | Specialized computational tool for factorization of descriptor contributions [1] |
| Octanol/Water Partition System | Reference system for lipophilicity measurements | Gold standard for log P determination [3] |
| Toluene/Water Partition System | Alternative partitioning system for HBD assessment | Surrogate for alkane/water system with better solubility properties [3] |
| PAMPA Assay Systems | High-throughput permeability screening | Various membrane compositions to mimic different biological barriers [1] |
Advanced Applications and Interpretation
Intermolecular Force Decomposition in Permeability Prediction
BR analysis provides critical insights into the dominant forces governing passive permeability through biological membranes [1]. The visualization of interaction forces enables researchers to select the most appropriate assay systems for specific drug discovery projects based on the balance of forces most relevant to their target tissue [1].
Diagram 2: Intermolecular Forces Governing Permeability
Quantitative Interpretation of BR Results
The output of BR analysis provides numerical and visual representation of the relative contribution of different descriptor blocks to lipophilicity and permeability models. The analysis of Δlog Poct–tol demonstrates that hydrogen bond donor properties account for the dominant share of variance in this parameter, confirming its utility as a specific measure of HBD capacity [3]. Similarly, when comparing permeability across different cell types, BR analysis reveals whether the same balance of intermolecular forces governs permeation in each system [1].
This interpretative framework enables medicinal chemists to make informed decisions about which molecular properties to modify for optimizing specific ADME parameters. For example, when poor permeability is identified as a limitation, BR analysis can determine whether the issue stems primarily from excessive hydrogen bond donation, molecular size, or other factors, directing the most efficient structural optimization strategy [1] [3].
Block Relevance analysis represents a significant advancement in the practical interpretation of lipophilicity data for drug discovery applications. By deconvoluting the complex balance of intermolecular forces governing partitioning and permeability phenomena, BR analysis enables medicinal chemists to select optimal measurement systems, interpret results mechanistically, and prioritize drug candidates based on a more sophisticated understanding of their physicochemical properties [1] [2]. The methodology bridges the gap between computational predictions and experimental measurements, providing a robust framework for optimizing compound properties throughout the drug development process.
The process of partitioning and chromatographic retention is governed by a complex interplay of various intermolecular forces. Deconvoluting these forces is essential for understanding and predicting molecular behavior in chemical and biological systems, particularly in drug discovery and development. The core principle involves breaking down the overall observed retention or partitioning phenomenon into its elementary interactions, such as hydrophobic effects, hydrogen bonding, and electrostatic forces. This quantitative dissection allows researchers to move beyond a simplistic, composite measurement like a chromatographic retention factor (log k) or a partition coefficient (log P) and gain a mechanistic understanding of the underlying physicochemical processes [4] [5].
This deconvolution is formally achieved through quantitative structure-property relationship (QSPR) modeling, often coupled with multivariate statistical methods like Partial Least Squares (PLS) regression [4]. The independent variables in these models are molecular descriptors that encode specific physicochemical properties. The Block Relevance (BR) analysis has emerged as a powerful computational tool built upon this framework, designed specifically to interpret these QSPR/PLS models and quantify the contribution of distinct blocks of intermolecular interactions to the overall property being studied [1] [5]. For lipophilicity assessment, this provides a nuanced picture that supports safer method selection and accelerates drug candidate prioritization [1].
Theoretical Framework and Key Concepts
Foundational Models for Deconvolution
The deconvolution of intermolecular forces rests on established theoretical models that relate a macroscopic property to a weighted sum of molecular interaction descriptors.
Linear Solvation Energy Relationships (LSER): This is a widely accepted model, often symbolized by the equation:
log k = c + eE + sS + aA + bB + vV[4]. In this equation, the capital letters represent solute properties (excess molar refractionE, dipolarity/polarizabilityS, hydrogen-bond acidityA, hydrogen-bond basicityB, and McGowan's characteristic volumeV), while the lower-case letters are system-specific coefficients that quantify the complementary interaction strength of the chromatographic system or partitioning environment [4]. Although powerful, a key limitation of the LSER approach is the reliance on empirical solvatochromic descriptors, which are not available for all compounds, particularly those with multiple functional groups or in ionized states [4].Molecular Interaction Fields (MIFs) and VolSurf+ Descriptors: To overcome the limitations of empirical descriptors, computational descriptors derived from Molecular Interaction Fields provide a robust alternative. A MIF is a 3D grid that maps the interaction energy between a target molecule and a chosen chemical probe (e.g., water, a dry probe, H-bond acceptor, H-bond donor) [4]. VolSurf+ is a computational tool that translates these complex 3D fields into a set of quantitative descriptors that characterize molecular size, shape, and interaction potential [4]. These descriptors are inherently interpretable, as they directly reflect a molecule's capability for specific interactions like hydrogen bonding and hydrophobic contact.
The Block Relevance (BR) Analysis
The BR analysis is a modern implementation that leverages the QSPR/PLS framework with VolSurf+-like descriptors [1] [5]. Its primary output is a visual and quantitative breakdown of the property (e.g., log k) into the following six fundamental blocks of intermolecular interactions:
- Size Block: Describes contributions from the size and shape of the solute molecule [5].
- DRY Block: Represents the hydrophobic interaction between the solute and the system, calculated using a "dry" (non-polar) probe [4] [5].
- O Block: Encodes the hydrogen-bond interaction where the solute acts as a hydrogen-bond donor (HBD) and the system (e.g., stationary phase) acts as a hydrogen-bond acceptor [4] [5].
- N1 Block: Encodes the hydrogen-bond interaction where the solute acts as a hydrogen-bond acceptor (HBA) and the system acts as a hydrogen-bond donor [4] [5].
- OH2 Block: Describes the solute's interaction with water, representing hydrophilicity [5].
- Others Block: Captures additional descriptors, often related to the imbalance between hydrophilic and hydrophobic regions on the molecular surface [4] [5].
The BR analysis produces plots that show the relative weight and direction (positive or negative contribution) of each block, providing an immediate visual assessment of the dominant forces governing a particular partitioning or retention process [5].
Logical Workflow of Force Deconvolution
The following diagram illustrates the conceptual and computational workflow for deconvoluting intermolecular forces using techniques like Block Relevance analysis.
Experimental Protocols
Protocol 1: Deconvoluting Chromatographic Retention Mechanisms
This protocol details the application of BR analysis to characterize the retention mechanism of a chromatographic system, such as the mixed-mode Celeris Arginine column [5].
- Objective: To dissect the individual contributions of intermolecular forces governing analyte retention on a chromatographic stationary phase.
- Materials and Reagents:
- HPLC system with UV detection or MS capability.
- Target analytical column (e.g., Celeris Arginine column).
- Test compounds (typically 50-100 structurally diverse solutes, including acids, bases, and neutrals).
- HPLC-grade solvents (water, acetonitrile).
- Buffer salts (e.g., ammonium acetate).
- Procedure:
- Sample Preparation: Prepare stock solutions of each test compound in a suitable solvent (e.g., DMSO) and further dilute with the mobile phase to working concentrations.
- Chromatographic Analysis:
- For each compound, measure the retention factor (log k) at multiple isocratic concentrations of organic modifier (e.g., from 10% to 90% v/v acetonitrile) [5].
- Maintain a constant buffer concentration (e.g., 10 mM ammonium acetate) and temperature (e.g., 25 °C) throughout.
- Ensure adequate equilibration time between mobile phase changes.
- Computational Processing:
- Data Analysis and Modeling:
- For each organic modifier percentage, build a PLS model with the measured log k values as the dependent variable (Y) and the VolSurf+ descriptors as independent variables (X).
- Input the PLS model and pre-defined descriptor blocks into the BR analysis software (e.g., implemented in MATLAB) [1].
- Interpret the resulting BR plots to identify the dominant interaction blocks (e.g., Size, O, DRY) and their relative contributions to retention at each solvent strength [5].
- Key Considerations: This approach is highly effective for neutral molecules. However, VolSurf+ descriptors have limitations for fully ionized compounds, which may require a complementary strategy using charge-based descriptors and Multiple Linear Regression (MLR) [5].
Protocol 2: High-Throughput Lipophilicity Measurement for Model Validation
This protocol describes a shake-flask method for the efficient experimental determination of distribution coefficients (log D) used to validate and ground computational models [6].
- Objective: To experimentally measure the lipophilicity (log D) of multiple compounds simultaneously, providing a high-throughput data source for QSPR model development and validation.
- Materials and Reagents:
- Research Reagent Solutions: See Table 1.
- LC-MS/MS system for analytical quantification.
- 96-well plates or individual test tubes.
- Phosphate buffer (pH 7.4) to simulate physiological conditions.
- 1-octanol (HPLC grade), pre-saturated with buffer.
- Procedure:
- Mixture Design: Combine up to 10 compounds into a single mixture, ensuring they are chromatographically separable and do not interact to cause ion pair partitioning artifacts [6].
- Partitioning:
- Add a known volume of the aqueous buffer (pre-saturated with 1-octanol) to the mixture of compounds.
- Add an equal volume of 1-octanol (pre-saturated with the buffer).
- Vortex-mix the samples vigorously for a set period (e.g., 1 hour) to reach partitioning equilibrium.
- Phase Separation: Centrifuge the samples to achieve complete phase separation.
- Quantification:
- Carefully separate the two phases.
- Dilute aliquots from both the aqueous and octanol phases as necessary.
- Analyze the diluted samples using LC-MS/MS to determine the concentration of each compound in both phases.
- Data Calculation:
- For each compound, calculate the distribution coefficient using the formula:
log D = log (Concentration_in_octanol / Concentration_in_aqueous)
- For each compound, calculate the distribution coefficient using the formula:
- Key Considerations: The use of compound mixtures significantly increases throughput. However, care must be taken to confirm the absence of intermolecular interactions that could skew the results, and the analytical method must be robust enough to quantify all compounds in the mixture accurately [6].
Data Presentation and Analysis
Research Reagent Solutions for Lipophilicity and Permeability Assessment
Table 1: Essential materials and reagents for experiments in lipophilicity assessment and permeability screening.
| Item | Function/Application |
|---|---|
| Celeris Arginine Column | A mixed-mode stationary phase used to study diverse interactions, particularly with anionic solutes; exemplary for method development and BR analysis [5]. |
| VolSurf+ Software | Generates molecular descriptors from 3D Molecular Interaction Fields (MIFs) for QSPR modeling; crucial for calculating interaction blocks in BR analysis [4]. |
| 1-Octanol & Buffer (pH 7.4) | The standard solvent system for shake-flask determination of distribution coefficients (log D), providing a gold-standard measure of lipophilicity [6]. |
| PAMPA (Parallel Artificial Membrane Permeability Assay) | A high-throughput screen for passive permeability; BR analysis can identify which PAMPA variant best mimics cellular permeability [1]. |
Table 2: Description of the fundamental interaction blocks used in Block Relevance analysis to deconvolute intermolecular forces [4] [5].
| Block Name | Physicochemical Interpretation | Role in Partitioning/Retention |
|---|---|---|
| Size | Molecular size and shape descriptors. | Governs cavity formation energy and dispersion interactions; can indicate a switch between reversed-phase and normal-phase chromatographic modes [5]. |
| DRY | Hydrophobic interactions quantified using a "dry" probe. | A primary driver in reversed-phase chromatography and octanol/water partitioning; a positive contribution increases retention with higher hydrophobicity [4]. |
| O (HBD) | Represents solute Hydrogen-Bond Donor ability. | Indicates the importance of the solute donating a hydrogen bond to the system; a negative contribution can reduce retention in a hydrophobic environment [4] [5]. |
| N1 (HBA) | Represents solute Hydrogen-Bond Acceptor ability. | Indicates the importance of the solute accepting a hydrogen bond from the system; interpreted relative to the O block [4] [5]. |
| OH2 | Hydrophilic interactions with water. | Describes the solute's affinity for the aqueous environment; a strong negative contribution favors partitioning into the aqueous phase [5]. |
| Others | Descriptors for imbalance of polar/apolar regions. | Captures additional complexity in molecular surfaces that is not fully described by the other blocks [4]. |
Experimental Data from a Model Chromatographic System
Table 3: Exemplary retention data (log k) for a series of drug-like compounds on the Celeris Arginine column at different concentrations of organic modifier (ACN) [5]. This data serves as the input (Y-variable) for building the QSPR/PLS models for BR analysis.
| Compound | Type | log k (10% ACN) | log k (20% ACN) | log k (50% ACN) | ... (other ACN%) |
|---|---|---|---|---|---|
| Compound A | Neutral | 1.45 | 0.98 | 0.15 | ... |
| Compound B | Acid | 2.10 | 1.65 | 0.45 | ... |
| Compound C | Base | 0.55 | 0.20 | -0.25 | ... |
| Compound D | Neutral | 1.20 | 0.75 | 0.05 | ... |
| ... | ... | ... | ... | ... | ... |
Note: The full dataset would typically include 50-100 compounds measured across 6-8 different percentages of organic modifier [5].
Application in Drug Discovery
The deconvolution of intermolecular forces via BR analysis provides critical, actionable insights in drug discovery. A key application is selecting the most relevant experimental methods for profiling compound properties.
For lipophilicity, BR analysis can identify which chromatographic system (e.g., specific stationary phase) best mimics the balance of intermolecular forces present in the standard 1-octanol/water system, thus providing a reliable and high-throughput log P surrogate [1]. Similarly, for permeability, it can determine which Parallel Artificial Membrane Permeability Assay (PAMPA) configuration replicates the passive transport mechanism seen in cellular models like Caco-2 [1]. This moves method selection from an empirical choice to a rational, mechanism-based decision, de-risking the use of surrogate assays and increasing the reliability of data used for candidate prioritization. Furthermore, understanding the specific interaction profile of a compound can guide medicinal chemists to make structural modifications that optimize the property balance, for instance, by reducing H-bond donor count to improve membrane permeability without disproportionately affecting target binding [1].
In modern drug discovery, the lipophilicity of a candidate compound, most often quantified as its n-octanol/water partition coefficient (log P), is a critical physicochemical parameter. It significantly influences a drug's absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties [7]. While the traditional method for determining log P is the shake-flask technique, reversed-phase high-performance liquid chromatography (RP-HPLC) has been widely adopted as an efficient, reproducible, and OECD-endorsed alternative [8] [7].
The core principle involves correlating a compound's chromatographic retention index (log k) with its known log P value using a set of standards, described by the Collander equation: log P = A log k + B [8]. However, a significant challenge arises because the relationship between log k and log P is not universal. The retention of a solute in a chromatographic system depends on a complex balance of intermolecular forces—such as hydrophobicity, hydrogen bonding, and ionic interactions—between the solute, the mobile phase, and the stationary phase [8] [2]. A log k value obtained from one chromatographic system cannot be directly converted to a true log P value if the system does not replicate the same balance of forces present in the octanol-water partitioning system.
This application note details the problem of converting chromatographic indices to log P and presents Block Relevance (BR) analysis as a solution to ensure the selection of appropriate, biomimetic chromatographic systems for reliable lipophilicity assessment.
The Core Scientific Challenge
The fundamental issue in converting log k to log P is that a single chromatographic index is a composite measure of all intermolecular interactions occurring in that specific system. The octanol-water system itself embodies a specific balance of these forces, which includes both hydrophobic effects and hydrogen-bonding capacity [2].
When a chromatographic system is used, the stationary phase (e.g., C18, IAM, cholesterol) and the mobile phase composition create a unique environment. If this environment does not mimic the solvation properties of the octanol-water system, the resulting log k will reflect a different balance of interactions, making direct conversion unreliable [8] [2]. For instance, a stationary phase that strongly engages in π-π or hydrogen-bonding interactions with a solute may yield a retention time that is not solely dependent on hydrophobicity, leading to an inaccurate log P estimation.
This challenge is particularly acute for ionizable compounds. While log P refers only to the neutral form, the distribution coefficient (log D) accounts for all ionized and unionized species at a specific pH, which is more physiologically relevant [7]. Chromatographic retention at a given pH is influenced by the ionization state of the analyte, and failure to account for this can lead to significant errors in predicting partitioning behavior [8].
Table 1: Key Challenges in Converting Chromatographic Indices to log P
| Challenge | Description | Impact on log P Prediction |
|---|---|---|
| Diverse Intermolecular Forces | Chromatographic retention is governed by hydrophobic, hydrogen-bonding, and ionic interactions. The relative contribution of each force varies with the stationary and mobile phases [8]. | If the chromatographic system's balance of forces differs from the octanol-water system, the correlation between log k and log P breaks down. |
| Stationary Phase Chemistry | Different phases (C18, IAM, C8, biphenyl) interact with solutes via different mechanisms. For example, IAM phases incorporate phospholipid groups to mimic cell membranes [8]. | A log k value from one phase cannot be directly compared to that from another, and a single universal conversion equation is not feasible. |
| Ionization of Analytics | At physiological pH (7.4), many drug-like compounds may be partially or fully ionized, affecting their retention behavior [8]. | Predictions based on retention times must use the distribution coefficient (log D) at the relevant pH, not the partition coefficient (log P) of the neutral form. |
| Validation of Correlation | A linear correlation between log k and log P must be established using standard compounds with known log P values for each specific chromatographic system. | Without a validated standard curve for a given system, the calculated log P or log D values are unreliable and not comparable to literature data. |
The Solution: Block Relevance (BR) Analysis
Block Relevance (BR) analysis is a computational tool implemented in MATLAB that deconvolutes the balance of intermolecular interactions governing a given phenomenon described by a QSPR/PLS model [1]. In the context of lipophilicity, BR analysis allows medicinal chemists to dissect the physicochemical nature of a chromatographic system and compare it directly to the octanol-water partitioning system.
The power of BR analysis lies in its ability to answer a critical question: "Does my chromatographic system express the same balance of solute-system forces as the octanol-water system?" [2]. By interpreting the output of a multivariate model, BR analysis identifies which blocks of molecular descriptors (e.g., those related to hydrophobicity, polarity, hydrogen-bonding) are most relevant for explaining the retention data in a given chromatographic system.
If the BR pattern of a chromatographic system matches that of the octanol-water system, then the interconversion of its retention index (log k) to log P is justified. This makes the choice of methods for measuring lipophilicity safer and speeds up the prioritization of drug candidates [1].
Experimental Protocols
Protocol 1: Determining Lipophilicity (log D7.4) Using a C-18 Column and a Standard Curve
This protocol outlines the steps to determine the distribution coefficient at pH 7.4 (log D7.4) using a standard C-18 column, as performed in studies of 1,3,4-thiadiazol derivatives [8].
4.1.1 Research Reagent Solutions
Table 2: Key Reagents and Materials for RP-HPLC log D Determination
| Item | Function / Description |
|---|---|
| Octadecyl (C-18) Column | Standard reversed-phase stationary phase; officially recognized by IUPAC and OECD for lipophilicity determination [8]. |
| Mobile Phase Buffer (pH 7.4) | A phosphate buffer is typically used to maintain physiological pH during analysis, critical for determining log D7.4 [8]. |
| Organic Modifier | Methanol or acetonitrile is used to adjust the eluting strength of the mobile phase [8]. |
| Standard Compounds | A set of compounds with known, reliably measured log P values, used to construct the standard curve [8]. |
4.1.2 Procedure
- Mobile Phase Preparation: Prepare a mobile phase consisting of a suitable buffer (e.g., phosphate) at pH 7.4 and an organic modifier (methanol or acetonitrile). Use isocratic conditions with a modifier concentration that provides adequate retention for the analytes.
- System Equilibration: Equilibrate the HPLC system with the mobile phase until a stable baseline is achieved.
- Void Time Determination: Inject an unretained compound (e.g., uracil or sodium nitrate) to determine the column's void time (t₀).
- Standard Analysis: Inject each standard compound and record its retention time (tᵣ). Calculate the capacity factor for each standard using the formula: log k = log[(tᵣ - t₀)/t₀].
- Calibration Curve: Construct a standard curve by plotting the known log P values of the standards against their experimentally determined log k values. Perform linear regression to obtain the equation log P = A log k + B.
- Analyte Measurement: Inject the compound of interest under identical conditions, determine its log k, and use the calibration equation to calculate its log D7.4.
Determining log D7.4 via RP-HPLC
Protocol 2: Evaluating Biomimetic Properties Using Specialized Stationary Phases
This protocol uses multiple stationary phases to gain a deeper understanding of a compound's lipophilicity and membrane permeability.
4.2.1 Research Reagent Solutions
Table 3: Specialized Stationary Phases for Biomimetic Chromatography
| Stationary Phase | Function / Description |
|---|---|
| Immobilized Artificial Membrane (IAM) | Silica surface modified with phosphatidylcholine groups. It mimics cell membranes and retention involves hydrophobic, ion-pairing, and H-bonding interactions [8]. |
| Chromatographic Hydrophobicity Index (CHI/IAM) | An extrapolated log kw parameter or index obtained from gradient elution on IAM columns [8]. |
| Cholesterol-Based Phase | Stationary phase with bonded cholesterol molecules; excellent for mimicking cellular membranes and predicting xenobiotic permeability [8]. |
| Biphenyl Phase | Retention is contributed by hydrophobic, π-π, steric, and hydrogen bond interactions, providing a complementary retention profile [8]. |
4.2.2 Procedure
- Column Selection: Select a set of different stationary phases (e.g., C-18, IAM, Cholesterol, Biphenyl).
- Isocratic Elution: For each column, run the compounds of interest under isocratic conditions with a mobile phase at pH 7.4.
- Data Collection: Determine the log kw (the extrapolated capacity factor for 0% organic modifier) for each compound on each stationary phase [8].
- Data Matrix Creation: Compile a data matrix where rows represent compounds and columns represent the different lipophilicity indices (log kC18, log kIAM, log k_CHOL, etc.).
- BR Analysis: Subject the data matrix to Block Relevance analysis. The BR analysis will deconvolute the balance of intermolecular forces driving retention in each system.
- System Comparison: Compare the BR pattern of each chromatographic system to that of the octanol-water system. A system with a similar BR profile is a good surrogate for log P.
Workflow for BR Analysis of Chromatographic Systems
Data Presentation and Analysis
The following table summarizes exemplary lipophilicity data obtained for a series of 1,3,4-thiadiazol-2-yl)-benzene-1,3-diols using various chromatographic systems, alongside computationally derived values [8].
Table 4: Exemplary Lipophilicity Data for 1,3,4-Thiadiazol Derivatives from Multiple Methods
| Compound | log k (C-18) | log D7.4 (C-18) | log k (IAM) | log k (Biphenyl) | In silico log P |
|---|---|---|---|---|---|
| 1 | 1.45 | 2.10 | 1.88 | 1.52 | 2.45 |
| 2 | 1.48 | 2.15 | 1.91 | 1.55 | 2.51 |
| 3 | 1.65 | 2.40 | 2.15 | 1.70 | 2.80 |
| 4 | 1.52 | 2.20 | 1.95 | 1.58 | 2.55 |
| ... | ... | ... | ... | ... | ... |
| Key Observation | A weak correlation was found between the experimental log k/log D and the in silico log P, highlighting the challenge of direct conversion and the potential differences in the underlying interaction balances [8]. |
Application of BR analysis to such a dataset would reveal, for instance, that retention on the IAM column is likely influenced more strongly by hydrogen-bonding descriptor blocks compared to the C-18 column. This objectively identifies the IAM system not as a pure hydrophobicity gauge, but as a superior biomimetic tool for predicting membrane penetration.
The conversion of chromatographic indices to log P is fraught with challenges stemming from the complex interplay of intermolecular forces in different systems. Simply assuming a direct correlation can lead to inaccurate and misleading lipophilicity estimates, jeopardizing critical decisions in drug candidate optimization.
Block Relevance analysis provides a powerful, rationale-driven solution to this problem. By enabling researchers to deconvolute the intermolecular interactions governing retention, BR analysis allows for the scientifically sound selection and validation of chromatographic systems that truly mimic the octanol-water partition process. Integrating BR analysis into the lipophilicity assessment workflow ensures more reliable data, enhances the efficiency of drug discovery, and provides deeper insights into the physicochemical properties that dictate a drug's fate in the body.
In modern drug discovery, predicting the absorption and permeability of candidate compounds is a critical challenge. Partial Least Squares (PLS) regression has emerged as a powerful chemometric tool for correlating the complex physicochemical properties of molecules with their biological behavior [9] [10]. When combined with advanced interpretation methods like Block Relevance (BR) analysis, researchers can deconvolute the balance of intermolecular forces governing pharmacokinetic properties, significantly accelerating candidate prioritization [1].
This Application Note provides detailed protocols for developing validated PLS models focused on permeability prediction and demonstrates how BR analysis enhances the interpretation of these models within lipophilicity assessment research. We frame these methodologies within the context of a broader thesis on BR analysis, highlighting its role in elucidating the mechanistic drivers of membrane permeability.
Theoretical Background
Partial Least Squares (PLS) in Pharmaceutical Sciences
PLS regression is a multivariate statistical technique particularly suited for situations where predictor variables (X) are numerous, collinear, and noisy. In QSAR/QSPR modeling, PLS effectively correlates a matrix of molecular descriptors (X-block) with biological activity or property data (Y-block) [9] [10]. The method projects the original variables into a reduced space of latent variables (components) that maximize the covariance between X and Y. A key strength of PLS is its ability to handle datasets where the number of variables exceeds the number of observations, a common scenario in pharmaceutical research where numerous molecular descriptors are calculated for a limited set of test compounds [9].
Block Relevance (BR) Analysis
BR analysis is a computational tool that extends the interpretability of PLS models. It works by grouping molecular descriptors into mechanistically meaningful blocks (e.g., size, polarity, hydrogen-bonding capacity) and then quantifies the relative contribution ("relevance") of each block to the overall model [1] [3]. This deconvolution allows medicinal chemists to move beyond black-box predictions and understand the fundamental intermolecular interactions—such as hydrophobic effects, hydrogen bonding, and steric constraints—that drive the property of interest [2]. For instance, BR analysis has proven instrumental in confirming that differences in partition coefficients (Δlog Poct–tol) are primarily governed by the hydrogen bond donor (HBD) capacity of solutes [3].
Application Protocol: Developing a PLS Model for Permeability Prediction
This protocol outlines the steps for constructing a PLS model to predict the apparent permeability coefficient (Papp) of drug candidates, based on a study of 33 steroids [9] [10].
Dataset Compilation and Curation
Objective: To assemble a high-quality dataset containing both experimental permeability data and calculated molecular descriptors.
Materials & Reagents:
- Test Compounds: A series of 33 steroids or relevant drug candidates.
- Software for Descriptor Calculation:
- Data Warrior: For predicting clogP, clogS, hydrogen bond acceptors/donors, and structural features [9].
- pkCSM Web Server: For predicting pharmacokinetic properties (Caco-2 permeability, intestinal absorption, etc.) from SMILES strings [9].
- Marvin by ChemAxon: For calculating pKa, logP, logD at pH 7.4, and logS [9].
- ACD/Labs Software: For obtaining molar volume, refractivity, and polarizability [9].
- PubChem Database: For accessing additional public data on molecular properties [9].
Procedure:
- Experimental Data Collection: Determine the apparent permeability coefficient (Papp) for each test compound using an appropriate in vitro system (e.g., Franz-type diffusion cells with a synthetic cellulose membrane) [9]. Record the Y-variable (e.g., Papp in cm/s).
- Descriptor Calculation (X-Variables): For each compound, calculate a wide array of physicochemical and structural descriptors. The original study utilized 37 such descriptors [9]. Key descriptors to include are:
- Lipophilicity Indices: logP (from multiple software programs for confirmation), logD at pH 7.4.
- Solubility: logS.
- Polarity and Hydrogen Bonding: Polar Surface Area (PSA), counts of hydrogen bond donors and acceptors.
- Size and Shape: Molecular weight (MW), molar volume, molar refractivity.
- Structural Features: Number of aromatic rings, rotatable bonds, rings, carbonyl groups, hydroxyl groups.
- Pharmacokinetic Predictors: Volume of distribution at steady state (VDss), Caco-2 permeability, skin permeability (log Kp) [9].
- Data Compilation: Construct a data matrix where rows represent the 33 compounds and columns represent the 37+ molecular descriptors (X-block) and the experimental Papp values (Y-block).
Model Construction and Validation
Software: Use specialized chemometric software such as Simca-P (Umetrics) or R/Python with PLS packages.
Procedure:
- Data Pre-processing: Normalize the X and Y variables using mean centering and unit variance scaling to ensure all descriptors contribute equally to the model [10].
- Model Training: Construct the PLS model by identifying latent variables that maximize the explanation of covariance between the X-block descriptors and the Y-block permeability data.
- Internal Validation: Perform cross-validation (e.g., 7-fold cross-validation) to assess the model's robustness and predictive ability [9] [10].
- Divide the data into 7 parts.
- Build a model using 6/7 of the data and predict the Y-values for the excluded 1/7.
- Repeat until every sample has been predicted once.
- Model Diagnostics: Evaluate the following key statistical parameters [9]:
- R²Y: The fraction of Y-variance explained by the model. A value >0.9 indicates excellent fit.
- Q²Y: The fraction of Y-variance predictable by the model, as estimated by cross-validation. A value >0.5 is considered good, and >0.7 is excellent.
- RMSEE: Root Mean Square Error of Estimation, indicating the model's fit error.
- RMSEP: Root Mean Square Error of Prediction, indicating the error in predicting new samples from cross-validation.
The workflow for the entire process, from data collection to model interpretation, is summarized in the diagram below.
Expected Results and Output
A robust PLS model for steroid permeability demonstrated the following performance metrics [9]:
- R²Y = 0.902
- Q²Y = 0.722
- RMSEE = 0.00265379
- RMSEP = 0.0077
Variable Importance: The Variable Importance in Projection (VIP) plot identified the following descriptors as most critical for predicting Papp [9]:
- logS (Water Solubility)
- logP (Partition Coefficient)
- logD (Distribution Coefficient)
- PSA (Polar Surface Area)
- VDss (Volume of Distribution at steady state)
Application Protocol: Interpreting PLS Models with Block Relevance Analysis
This protocol describes how to use BR analysis to extract mechanistic insights from a validated PLS model.
Objective: To deconvolute the balance of intermolecular interactions captured by a PLS model, thereby facilitating a more chemically intuitive interpretation.
Software: The BR analysis can be performed using its implementation in MATLAB or other computational environments [1].
Procedure:
- Descriptor Blocking: Group the molecular descriptors from the PLS model into a few (e.g., 4-6) mechanistically meaningful blocks. A suggested grouping is:
- Size/Shape Block: Descriptors like molecular weight, molar volume, surface area.
- Lipophilicity Block: logP, logD.
- Hydrogen Bonding Block: PSA, counts of H-bond donors and acceptors, calculated logS.
- Polarity/Polarizability Block: Molar refractivity, polarizability.
- BR Analysis Execution: Process the PLS model and the predefined descriptor blocks using the BR analysis algorithm.
- Interpretation of Results: The analysis will output the relevance of each block to the model. A higher relevance value indicates that the intermolecular interactions represented by that block are more critical in determining the property (e.g., permeability).
Case Study: Interpretation of Δlog Poct–tol
In a study investigating the difference between octanol/water and toluene/water partition coefficients (Δlog Poct–tol), BR analysis was applied to a PLS model built with 82 VolSurf+ descriptors. The descriptors were grouped into six blocks. The analysis clearly showed that the hydrogen bond donor (HBD) properties block was the most relevant, demonstrating that Δlog Poct–tol is a clean descriptor for a solute's HBD capacity [3]. This insight is crucial for assessing a molecule's potential to form intramolecular hydrogen bonds, a key factor in membrane permeability.
The conceptual process of the BR analysis is as follows.
The selection of appropriate molecular descriptors is fundamental to building a predictive PLS model. The table below summarizes the key descriptor categories and their roles in permeability modeling.
Table 1: Essential Physicochemical Descriptors for Permeability PLS Models
| Descriptor Category | Specific Descriptors | Role in Permeability Prediction | Software/Tool for Calculation |
|---|---|---|---|
| Lipophilicity | logP, logD (at pH 7.4) | Models passive diffusion through lipid membranes; optimal logP is often critical. | Marvin, Data Warrior [9] |
| Solubility | logS | Reflects the compound's aqueous solubility, a prerequisite for absorption. | Marvin, Data Warrior [9] |
| Size & Bulkiness | Molecular Weight (MW), Molar Volume, Molar Refractivity | Larger molecules may have restricted diffusion through membrane pores. | ACD/Labs, Data Warrior [9] |
| Polarity & H-Bonding | Polar Surface Area (PSA), H-Bond Donors, H-Bond Acceptors | High polarity and H-bonding potential can reduce permeation by desolvation penalties. | Data Warrior, PubChem [9] |
| Structural Features | Number of Rotatable Bonds, Rings, Aromatic Rings | Related to molecular flexibility and rigidity, influencing conformation during permeation. | Data Warrior, Marvin [9] |
| Distribution | VDss (Volume of Distribution) | A pharmacokinetic parameter correlated with tissue permeability and binding. | pkCSM [9] |
Research Reagent Solutions
Table 2: Essential Research Reagents and Software Tools
| Item | Function/Application | Example/Supplier |
|---|---|---|
| Franz Diffusion Cell System | In vitro apparatus for measuring permeation rates of compounds across artificial or biological membranes. | Standard vertical Franz cells [9] |
| Artificial Membranes | Synthetic models (e.g., cellulose) for high-throughput permeability screening. | Synthetic cellulose membrane [9] |
| Cheminformatics Software | Calculates 2D and 3D molecular descriptors from chemical structures. | Data Warrior, Marvin, ACD/Labs [9] |
| Online Prediction Servers | Provides predictions for ADMET and physicochemical properties from SMILES notation. | pkCSM [9] |
| Multivariate Analysis Software | Constructs, validates, and interprets PLS models and other multivariate statistical analyses. | Simca-P (Umetrics) [9] [10] |
| BR Analysis Tool | A computational tool for deconvoluting and interpreting PLS models. | MATLAB implementation [1] |
Troubleshooting and Technical Notes
- Low Predictive Power (Q²Y): This can result from a high degree of non-linearity in the data or insufficiently relevant descriptors. Consider non-linear PLS variants, ensure the dataset is well-curated, and expand the descriptor set to capture more diverse molecular features.
- Overfitting: Indicated by a high R²Y but a low Q²Y. Mitigate this by using cross-validation to select the optimal number of latent components and avoiding an excessive number of components relative to the number of observations.
- Ambiguous Interpretation from VIP Scores: While VIP scores identify important descriptors, they do not reveal the underlying intermolecular forces. This is the specific problem that BR analysis solves by grouping descriptors and quantifying the relevance of each mechanistic block [1] [3].
- Applicability Domain: Always define the chemical space of the training set. Predictions for compounds structurally dissimilar to the training set are unreliable. Use leverage and distance-to-model plots to identify such outliers.
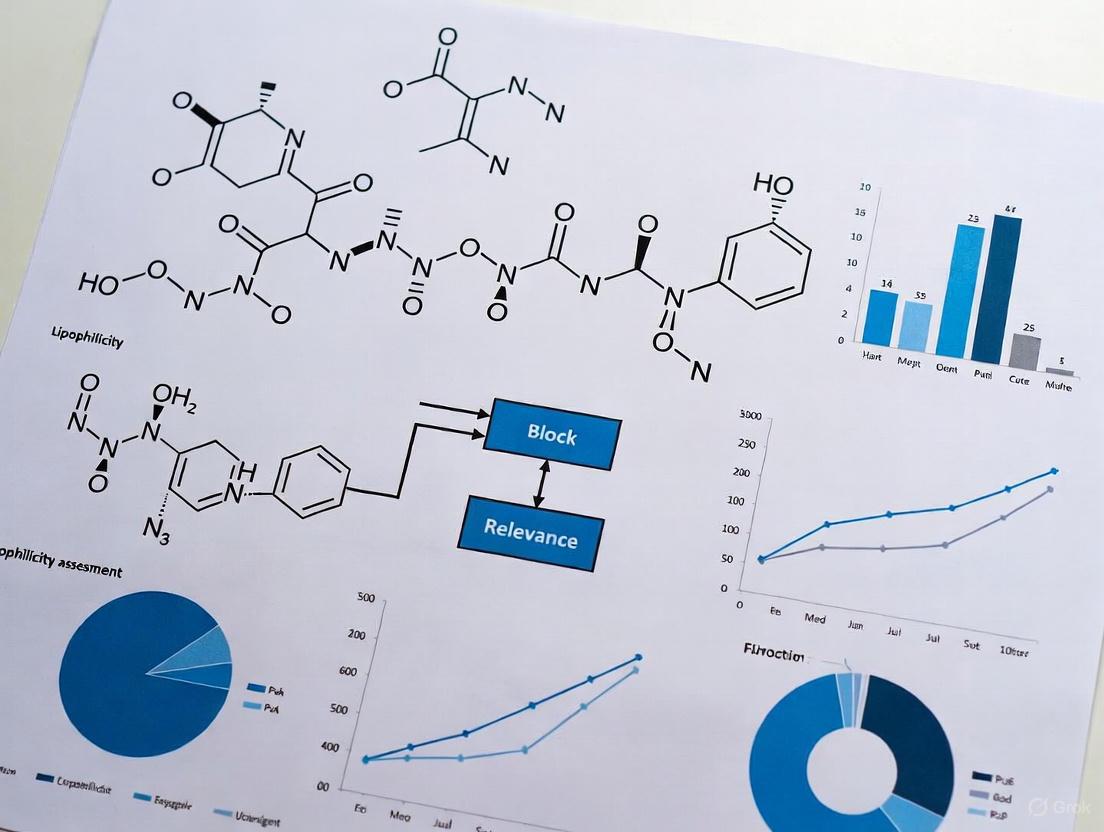
Block Relevance analysis in lipophilicity assessment research provides a framework for deconstructing complex molecular structures into functionally distinct subunits, or "blocks," to quantify their individual contributions to a molecule's overall physicochemical behavior. A critical step in this analytical process is the effective visual representation of these blocks and their complex interactions. Proper graphical representation transforms abstract numerical data into an intuitive format, enabling researchers to instantly identify key structure-activity relationships and the dominant blocks governing lipophilicity.
This document outlines standardized protocols for creating clear, accessible, and scientifically rigorous visualizations of Interaction Blocks, specifically tailored for lipophilicity assessment and related drug discovery applications. The methodologies described herein integrate current best practices in data visualization and scientific communication to ensure that the resulting graphics are not only visually compelling but also analytically precise and accessible to a diverse scientific audience, including those with color vision deficiencies [11] [12].
Table 1: Experimental Lipophilicity Parameters for a Series of Tacrine-Based Compounds
This table summarizes key chromatographic parameters used to determine lipophilicity, which serve as foundational data for Block Relevance analysis. The values R_M^0 and C_0 are derived from reversed-phase thin-layer chromatography (RP-TLC) and represent a reliable measure of compound lipophilicity [13].
| Compound ID | Piperidine Substituent | R_M^0 (MeOH) | C_0 (MeOH) | % Plasma Protein Binding (PPB) |
|---|---|---|---|---|
| 1 | Phenyl | 1.45 | 0.32 | 82.38% |
| 2 | Nicotinoyl | 1.21 | 0.41 | 85.65% |
| 3 | Nicotinoyl | 1.33 | 0.38 | 87.92% |
| 4 | Nicotinoyl | 1.28 | 0.39 | 84.29% |
| 5 | 2-Chlorophenyl | 1.89 | 0.25 | 92.47% |
| 6 | 3-Chlorophenyl | 1.76 | 0.27 | 90.15% |
| 7 | 4-Chlorophenyl | 1.81 | 0.26 | 94.54% |
| 8 | 2-Fluorophenyl | 1.59 | 0.29 | 89.01% |
| 9 | 3-Fluorophenyl | 1.52 | 0.30 | 87.33% |
| 10 | 4-Fluorophenyl | 1.55 | 0.29 | 91.22% |
| 11 | 2-Methoxyphenyl | 1.49 | 0.31 | 86.74% |
| 12 | 3-Methoxyphenyl | 1.41 | 0.33 | 85.90% |
| 13 | 4-Methoxyphenyl | 1.44 | 0.32 | 98.16% |
Table 2: Computed Interaction Descriptors for Block Relevance Analysis This table lists common computational descriptors used to characterize the properties of individual molecular blocks and their potential interactions. These parameters are essential for building predictive models of lipophilicity and plasma protein binding [14] [13].
| Descriptor Category | Specific Descriptor | Relevance to Lipophilicity & Block Analysis |
|---|---|---|
| Hydrophobicity | LogP | Measures overall partition coefficient; primary target for Block Relevance analysis. |
| Topological | Polar Surface Area (PSA) | Indicates a block's capacity for hydrogen bonding, negatively correlating with passive membrane permeability. |
| Electronic | Partial Charges | Determines a block's potential for electrostatic interactions with proteins or phospholipids. |
| Steric | Molar Refractivity | Represents the volume and polarizability of a block, influencing van der Waals forces. |
| Structural | Presence of Aromatic Rings | Key for π-π stacking interactions with proteins like Human Serum Albumin (HSA). |
Visualization Framework for Interaction Blocks
Effective visualization is paramount for interpreting Block Relevance analysis. The following diagrams, created using Graphviz DOT language, provide standardized templates for representing the analytical workflow and the interaction of molecular blocks with their biological targets. The color palette and design principles adhere to accessibility guidelines, ensuring clarity for all readers [11] [15].
Workflow for Lipophilicity Assessment and Block Analysis
This diagram outlines the integrated experimental and computational pipeline for determining lipophilicity and performing a Block Relevance analysis.
Molecular Block Interaction with Human Serum Albumin
This diagram conceptualizes the binding of a tacrine-based molecule (divided into its core and substituent blocks) to the Sudlow site I of Human Serum Albumin (HSA), a key plasma protein.
Experimental Protocols
Protocol 1: Determination of Lipophilicity using Reversed-Phase Thin-Layer Chromatography (RP-TLC)
Principle: Lipophilicity is assessed by measuring the retention behavior of compounds on a hydrophobic stationary phase using mobile phases of varying polarity. The derived parameters (R_M^0 and C_0) are highly correlated with the octanol-water partition coefficient (LogP) [13].
Materials:
- Stationary Phase: TLC plates pre-coated with RP-18 F254s silica gel.
- Mobile Phase: Binary mixtures of an organic modifier (e.g., Methanol, Acetonitrile) and water, acidified with 0.1% formic acid (v/v). Prepare a series of mobile phases with organic modifier concentrations (e.g., 50%, 60%, 70%, 80%, 90% v/v).
- Sample Solutions: Dissolve test compounds in a suitable solvent (e.g., methanol) to a concentration of ~0.5 mg/mL.
- Equipment: A vertical developing chamber, a UV lamp (λ = 254 nm), and a micropipette.
Procedure:
- Spotting: Using a micropipette, apply 1.0 μL of each sample solution onto the baseline of the RP-TLC plate. Allow spots to dry completely.
- Development: Pour the mobile phase into the development chamber to a depth of about 1 cm and saturate the atmosphere for 30 minutes. Place the spotted plate into the chamber and allow the mobile phase to develop until the solvent front is near the top of the plate (~8-9 cm).
- Drying & Visualization: Remove the plate from the chamber, mark the solvent front, and allow it to air-dry in a fume hood. Visualize the separated spots under UV light at 254 nm.
- Data Measurement: Measure the distance from the start point to the center of the spot (compound migration distance,
Z_x) and from the start point to the solvent front (Z_f). - Calculation: Calculate the retention factor,
R_M, for each mobile phase composition.R_f = Z_x / Z_fR_M = log(1/R_f - 1)
- Lipophilicity Parameters: Plot
R_Mvalues against the volume fraction of the organic modifier (φ). The y-intercept of the linear regression isR_M^0, a direct measure of lipophilicity. The slope provides information about the specific hydrophobic surface area. TheC_0value, calculated as the negative ratio of the intercept to the slope, is also a validated lipophilicity parameter.
Protocol 2: High Performance Affinity Chromatography (HPAC) for Plasma Protein Binding
Principle: This method uses a chromatography column with immobilized Human Serum Albumin (HSA) to mimic the drug-protein binding that occurs in plasma. Compounds with higher affinity for HSA will have longer retention times on the column [13].
Materials:
- HPAC Column: A commercially available column (e.g., 50 mm x 4.0 mm) with HSA chemically bonded to silica particles.
- Mobile Phase: Phosphate buffer (e.g., 50 mM, pH 7.4) with a gradient of 2-propanol (e.g., 0-10% v/v).
- HPLC System: A standard HPLC system equipped with a pump, autosampler, column oven, and UV/VIS or DAD detector.
- Reference Compounds: A set of drugs with known HSA binding percentages (e.g., Warfarin) for calibration.
Procedure:
- System Equilibration: Install the HSA column and equilibrate it with the initial mobile phase (e.g., phosphate buffer with 0% 2-propanol) at a constant flow rate (e.g., 0.5 mL/min) until a stable baseline is achieved.
- Calibration: Inject reference compounds with known low, medium, and high HSA binding. Record their retention times. Establish a correlation between retention time (or capacity factor k') and literature PPB values.
- Sample Analysis: Inject the test compound solution. Use an isocratic or gradient elution method. A typical mobile phase is phosphate buffer (pH 7.4) and 2-propanol.
- Data Analysis: Record the retention time of the test compound. Calculate the capacity factor,
k' = (t_R - t_0)/t_0, wheret_Ris the compound's retention time andt_0is the column void time. - Binding Affinity Estimation: Compare the
k'value of the test compound to the calibration curve established from reference compounds to estimate its plasma protein binding affinity.
Protocol 3: Computational Docking to Analyze Block-Specific Protein Interactions
Principle: Molecular docking simulations predict the preferred orientation of a molecule (divided into its constituent blocks) when bound to a target protein like HSA. This provides atom-level insight into the forces (hydrogen bonds, hydrophobic, π-π stacking) driving the interaction for each block [14] [13].
Materials:
- Software: A molecular docking program (e.g., AutoDock Vina, GOLD, Schrödinger Glide).
- Protein Structure: The 3D crystal structure of the target protein (e.g., HSA, PDB ID: 1AO6) from the Protein Data Bank.
- Ligand Structures: 3D chemical structures of the molecules to be docked, in a suitable file format (e.g., .mol2, .sdf).
Procedure:
- Protein Preparation: Download and prepare the protein structure. This involves adding hydrogen atoms, assigning partial charges, and defining the binding site (e.g., Sudlow site I for HSA based on literature).
- Ligand Preparation: Draw or obtain the 3D structures of the test compounds. Perform energy minimization to optimize their geometry. Define the rotatable bonds.
- Docking Execution: Run the docking simulation for each ligand against the prepared protein. Set the search space to encompass the entire defined binding site.
- Analysis of Results: Analyze the top-ranked docking poses. Identify specific interactions (hydrogen bonds, hydrophobic contacts, π-π stacking, salt bridges) between atoms in each defined molecular block (e.g., tacrine core vs. substituent) and the amino acid residues in the protein binding pocket.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Lipophilicity and Interaction Analysis
| Category | Item / Reagent | Function & Application Notes |
|---|---|---|
| Chromatography | RP-TLC Plates (RP-18 F254s) | Hydrophobic stationary phase for experimental lipophilicity determination via RP-TLC [13]. |
| HSA-HPAC Column | Immobilized protein stationary phase for high-throughput assessment of plasma protein binding affinity [13]. | |
| Methanol & Acetonitrile (HPLC Grade) | Organic modifiers for creating mobile phases of varying polarity in chromatographic separations. | |
| Computational Tools | Graph Neural Networks (GNNs) | Explicitly learns molecular structure from graph representations to predict drug-target interactions and properties [14]. |
| Transformer Models | Uses self-attention mechanisms on SMILES strings to implicitly capture structural context and predict bioactivity [14]. | |
| Docking Software (e.g., AutoDock Vina) | Predicts binding orientation and affinity of molecular blocks to protein targets like HSA [13]. | |
| Visualization & Analysis | Adobe Color | Online tool for designing and validating color-blind safe palettes for scientific figures [12]. |
| Colour Contrast Analyser (CCA) | Software tool for verifying that color contrast ratios in diagrams meet WCAG accessibility standards (≥ 4.5:1 for text) [11] [15]. | |
| Graphviz (DOT language) | Open-source tool for creating precise, script-based diagrams of workflows and interaction networks, ensuring reproducibility. |
Practical Applications of BR Analysis in Lipophilicity and Permeability Assessment
Selecting Optimal Chromatographic Systems as log P Surrogates
Within drug discovery, accurate assessment of lipophilicity is paramount, as this key physicochemical property significantly influences a compound's absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile. The traditional measure of lipophilicity, the octanol-water partition coefficient (log P), often fails to accurately predict membrane permeability and other biological partitioning processes because the octanol-water system does not fully capture the balance of intermolecular forces present in biological environments [16]. Block Relevance (BR) analysis has emerged as a powerful computational tool that deconvolutes the balance of intermolecular interactions governing a drug discovery-related phenomenon described by a QSPR/PLS model [1]. This application note details how BR analysis guides the selection and validation of optimal chromatographic systems as reliable surrogates for biologically relevant lipophilicity measurements, thereby accelerating drug candidate prioritization.
Theoretical Foundation: Block Relevance Analysis
Block Relevance analysis, implemented in MATLAB, provides a framework for interpreting complex partitioning and retention phenomena in a manner accessible to medicinal chemists [2]. The core principle of BR analysis involves deconstructing a given physicochemical or biological process into its fundamental interaction blocks, typically comprising:
- Van der Waals interactions (dispersion forces)
- Hydrogen-bonding donor acidity
- Hydrogen-bonding acceptor basicity
- Dipole-dipole and electrostatic interactions
By quantifying the contribution of each interaction block, BR analysis enables direct comparison between different partitioning systems, such as chromatographic retention and biological membrane permeability [1] [2]. When the interaction balance of a chromatographic system closely mirrors that of a target biological process (e.g., passive permeability through cellular membranes), that system can serve as a predictive surrogate, reducing the need for more complex and resource-intensive biological assays.
Selecting Chromatographic Systems as log P Surrogates
System Comparison Using Abraham Solvation Parameters
A robust approach for identifying surrogate chromatographic systems involves characterizing both the biological and chromatographic systems using Abraham's solvation model [17]. This model describes interactions using system constants (e, s, a, b, v) representing excess electron lone pair interactions, dipole-type interactions, hydrogen-bond basicity, hydrogen-bond acidity, and dispersion interactions, respectively.
The similarity between two systems can be quantified by calculating the Euclidean distance between their system constant vectors in the five-dimensional parameter space [17] [18]: Distance = √[(e₁ - e₂)² + (s₁ - s₂)² + (a₁ - a₂)² + (b₁ - b₂)² + (v₁ - v₂)²]
A smaller Euclidean distance indicates greater similarity between the chromatographic system and the biological process, suggesting its potential as an effective surrogate.
Chromatographic Systems for Specific Applications
Table 1: Optimal Chromatographic Surrogates for Various Biological Processes
| Biological Process | Recommended Chromatographic System | Key Similarities | Performance Metrics |
|---|---|---|---|
| Non-specific toxicity (Fathead minnow) | IAM PC DD 2 column; 10% methanol-water mobile phase [18] | Balanced hydrogen bonding and dispersion interactions | Standard Error: 0.22 log units; R² = 0.97 [18] |
| Soil-water distribution | Bakerbond DIOL column; 20% acetonitrile-water mobile phase [18] | Emphasis on hydrophobic and weak polar interactions | Standard Error: 0.38 log units; R² = 0.88 [18] |
| Passive membrane permeability (bRo5 compounds) | Polystyrene-divinylbenzene matrix (PRP-C18); isocratic 60% organic phase [16] | Sensitivity to H-bond donor desolvation penalty; purely apolar environment | R² = 0.97 for predicting Log Ddd/w; RMSD = 0.356 [16] |
| Hydrocarbon-water partitioning | Pure polystyrene-divinylbenzene matrix [16] | Purely apolar environment mimicking hydrocarbon solvents | Strong correlation with toluene-water partition coefficients [16] |
Experimental Protocols
Protocol 1: BR-Assisted Method Selection for Reliable log PoctSurrogates
Principle: Identify chromatographic conditions that best emulate the intermolecular interaction balance of the octanol-water system [1].
Materials:
- HPLC system with variable UV detector
- Columns with different stationary phases (C18, IAM, PRP-C18, etc.)
- Mobile phases: methanol-water and acetonitrile-water mixtures
- Reference compounds with known log Poct values
Procedure:
- Characterize the octanol-water system using Abraham's model by analyzing the retention of reference compounds with diverse functionalities.
- Characterize candidate chromatographic systems using the same set of reference compounds under various mobile phase compositions.
- Perform BR analysis to determine the contribution of each interaction block for both the octanol-water and chromatographic systems.
- Calculate Euclidean distances between the octanol-water system and each chromatographic system based on their Abraham system constants.
- Select the optimal system with the smallest Euclidean distance (typically ≤0.2 difference in system constant ratios) [18].
- Validate the surrogate by measuring the retention factors of test compounds and correlating with their reference log Poct values.
Protocol 2: High-Throughput Chromatographic log P Determination
Principle: Utilize reverse-phase HPLC to measure log P values for common drugs, providing a robust alternative to traditional shake-flask methods [19].
Materials:
- RP-HPLC system with C18 column
- Phosphate buffers (pH 6.0 and 9.0)
- Reference standards with well-established log P values
- Test compounds (e.g., rivaroxaban, carbamazepine, ibuprofen)
Procedure:
- Prepare mobile phases using pH 6.0 and pH 9.0 buffers mixed with acetonitrile or methanol.
- Generate calibration curves at each pH using reference standards with known log P values.
- Inject test compounds and measure retention times under isocratic conditions.
- Calculate capacity factors (k) from retention times.
- Determine log P values by interpolation from the calibration curves.
- Report results as the average of values obtained at both pH levels.
Protocol 3: Chromatographic Measurement of Permeability-Relevant Lipophilicity for bRo5 Compounds
Principle: Estimate hydrocarbon-water partition coefficients for macrocyclic peptides and other beyond Rule of 5 (bRo5) compounds using a chromatographic approach [16].
Materials:
- HPLC system with PRP-C18 column or silica-C18 column
- Mobile phase: acetonitrile-phosphate buffered saline (PBS) mixtures
- 1,9-decadiene for shake-flask reference measurements
Procedure:
- Condition the column (PRP-C18 recommended) with mobile phase.
- Employ isocratic elution with 60% organic phase or a gradient from 20-100% organic phase.
- Measure retention times for training set compounds with known Log Ddd/w (1,9-decadiene-water distribution coefficients).
- Calculate capacity factors (LogK') from retention times.
- Establish correlation model using exponential fit: Log EDdd/w = 2.26 × (1 - e(-1.40 × LogK'60)) [16].
- Apply the model to predict Log Ddd/w for test compounds from their chromatographic retention.
Workflow Visualization
Workflow for Selecting log P Surrogates
The Scientist's Toolkit
Table 2: Essential Research Reagents and Materials
| Item | Function/Application | Key Characteristics |
|---|---|---|
| IAM PC DD 2 Column | Surrogation of biological membrane partitioning [18] | Immobilized artificial membrane phosphatidylcholine analogues |
| PRP-C18 Column | Hydrocarbon-water partitioning surrogate for bRo5 compounds [16] | Pure polystyrene-divinylbenzene matrix; no silanol groups |
| Supelcosil LC-ABZ Column | General lipophilicity screening with balanced interactions [2] | C18/amide-based stationary phase with low silanol activity |
| Reference Standard Set | System characterization and calibration [19] [17] | Compounds with known log P spanning diverse functionalities |
| Polystyrene-divinylbenzene Matrix | Measuring permeability-relevant lipophilicity [16] | Purely apolar environment mimicking hydrocarbon solvents |
The strategic selection of chromatographic systems as log P surrogates, guided by Block Relevance analysis, represents a significant advancement in lipophilicity assessment for drug discovery. By systematically matching the interaction balance of chromatographic systems to target biological processes, researchers can implement predictive, high-throughput screening methods that more accurately forecast compound behavior in biological systems. The protocols outlined herein provide practical frameworks for deploying these approaches, enabling more efficient and informed decision-making in drug candidate prioritization.
Lipophilicity, the affinity of a molecule for a lipophilic environment versus an aqueous one, is a fundamental physicochemical property in drug discovery. It profoundly influences a compound's absorption, distribution, metabolism, excretion, and toxicity (ADMET). Characterizing lipophilicity becomes particularly challenging for amphoteric and zwitterionic compounds, which contain both acidic and basic functional groups. These compounds can exist in multiple ionization states, including a neutral zwitterionic form that bears both a positive and a negative charge, depending on the pH of their environment [20].
The lipophilicity-pH profiling of these molecules is complex yet critical, as common drugs like antibacterials, antiallergics, and diuretics often fall into this category [20]. Traditional lipophilicity measurements, such as the shake-flask method, can be complicated by drug-buffer interactions and ion-pairing effects [20]. Furthermore, a new class of "nonclassical zwitterions" has been identified. These molecules exhibit zwitterionic behavior despite a low or negative ΔpKa (pKa basic - pKa acidic), often characterized by weak acidic and basic pKa values and conjugation through an extended aromatic system [21] [22]. In contrast to most classical zwitterions, nonclassical zwitterions can combine excellent permeability with low lipophilicity, making them an attractive design principle in medicinal chemistry [21] [22].
This Application Note details practical methodologies for the accurate determination and interpretation of lipophilicity for zwitterionic compounds, framing these techniques within the advanced data analysis framework of Block Relevance (BR) analysis. BR analysis is a computational tool that deconvolutes the balance of intermolecular interactions governing a given drug discovery-related phenomenon described by a QSPR/PLS model, thereby aiding in method selection and data interpretation [1] [2].
Experimental Determination of Lipophilicity for Zwitterions
For zwitterionic compounds, lipophilicity is not a single value but a profile across a physiological pH range. Several experimental techniques are available, each with distinct advantages and limitations.
Comparative Analysis of Key Methods
The table below summarizes the core methodologies for profiling zwitterion lipophilicity.
Table 1: Comparison of Key Methods for Lipophilicity Profiling of Zwitterionic Compounds
| Method | Principle | Key Advantages | Key Limitations | Suitability for Zwitterions |
|---|---|---|---|---|
| Shake-Flask (Octanol/Water) [20] | Direct partitioning of the drug between 1-octanol and aqueous buffer, followed by concentration analysis. | Considered a gold standard; provides direct measurement. | Time-consuming; potential for drug-buffer interactions; requires compound-specific analytical methods. | High, but ion-pairing with buffer components can affect results for charged species. |
| Potentiometric Titration [20] | Two-phase titration where the partition coefficient is calculated from the shift in pKa upon addition of the water-immiscible phase (e.g., octanol). | Avoids different ion-pairing buffers; lower determination times; provides pKa values simultaneously. | Requires specialized equipment (pH meter, autotitrator). | Highly suitable; leads to similar log D profiles as shake-flask but is often more convenient. |
| Chromatographic Hydrophobicity Index (CHI) [20] | Uses reversed-phase HPLC retention time with a fast gradient to derive a high-throughput lipophilicity index. | Rapid and efficient for screening; requires minimal compound. | May differ slightly from direct partitioning methods; requires calibration. | Excellent for high-throughput screening and profiling across pH (CHI vs. pH). |
| Δlog P Oct-Tol [3] | Difference between log P in octanol/water and log P in toluene/water systems. | Serves as a molecular descriptor for solute hydrogen bond donor (HBD) properties and propensity for intramolecular hydrogen bonds (IMHB). | Requires two separate log P measurements. | Powerful for interpreting the behavior of zwitterions, particularly nonclassical ones with IMHB. |
Detailed Protocols
Protocol 1: Shake-Flask Method for log D Determination
This protocol is adapted from established procedures in the literature [20].
1. Research Reagent Solutions:
- Lipophilic Phase: 1-Octanol (saturated with aqueous buffer).
- Aqueous Phase: Aqueous buffer (e.g., phosphate, citrate) at desired pH, saturated with 1-octanol.
- Standard Solutions: Stock solution of the zwitterionic test compound in a suitable solvent (e.g., DMSO, methanol).
2. Procedure: 1. Prepare the pre-saturated octanol and buffer phases by mixing them overnight on a magnetic stirrer and allowing them to separate. 2. Add a known volume of the aqueous phase (e.g., 2 mL) to a suitable vial or tube. 3. Spike a known, small volume of the compound stock solution into the aqueous phase to avoid significant solvent effects. 4. Add an equal volume of the octanol phase to the vial. 5. Seal the vial and shake it vigorously for a sufficient time to reach equilibrium (e.g., 1 hour at constant temperature). 6. Centrifuge the mixture to achieve complete phase separation. 7. Carefully separate the two phases and analyze the concentration of the compound in each phase using a compound-specific method (e.g., HPLC-UV, LC-MS). 8. Include a control without the organic phase to check for compound adsorption to the vial.
3. Data Analysis:
Calculate the distribution coefficient, D, at the specific pH using the formula:
D = (Concentration in octanol phase) / (Concentration in aqueous phase)
log D = log (Concentration in octanol / Concentration in aqueous)
A full lipophilicity profile is generated by repeating this procedure across a relevant pH range (e.g., 2-12).
Protocol 2: Potentiometric log D Determination
This protocol is based on the pH-metric technique [20].
1. Research Reagent Solutions:
- Titrants: Standardized acid (e.g., 0.5 M HCl) and base (e.g., 0.5 M KOH).
- Ionic Strength Adjuster: 0.15 M KCl solution to maintain a constant ionic strength.
- Lipophilic Phase: 1-Octanol.
2. Procedure: 1. Dissolve the zwitterionic compound in a water-miscible solvent (if necessary) and add it to a vessel containing the KCl solution. 2. Titrate the aqueous solution under an inert atmosphere (e.g., N₂) to obtain the pKa values of the compound in the aqueous phase. 3. Add a known volume of octanol to the system and titrate again. The presence of the octanol phase will cause a shift in the apparent pKa values as the compound partitions. 4. Perform the titration over a wide pH range, ensuring the compound remains stable.
3. Data Analysis: Specialized software (e.g., pDISOL-X, Sirius) is used to analyze the titration curves from the aqueous-only and two-phase experiments. The software calculates the partition coefficient for the neutral species (log P) and the distribution coefficients (log D) at any pH based on the observed pKa shifts.
Protocol 3: Determination of CHI and log k'80
This protocol describes a high-throughput chromatographic approach [20].
1. Research Reagent Solutions:
- Mobile Phase A: Aqueous buffer (e.g., 50 mM ammonium acetate, pH 7.4).
- Mobile Phase B: Organic modifier (e.g., Acetonitrile).
- Column: Reversed-phase column (e.g., C18, or Supelcosil LC-ABZ for higher versatility [2]).
2. Procedure: 1. Set up a fast gradient run, for example, from 5% to 100% B over 5-10 minutes. 2. Inject the test compound and record its retention time. 3. Inject a set of standards with known CHI values (e.g., a cocktail of drugs) to calibrate the system.
3. Data Analysis: The Chromatographic Hydrophobicity Index (CHI) is calculated from the gradient retention time and the calibration curve. This value can be used directly as a lipophilicity index or converted to a log P scale. For a more direct measure of lipophilicity in a nonpolar environment, an isocratic method with a high organic modifier content (e.g., log k'80 PLRP-S) can be used [23].
Interpreting Data with Block Relevance (BR) Analysis
Once experimental data is acquired, BR analysis provides a powerful framework for interpretation, moving beyond a single lipophilicity value to understand the underlying intermolecular forces.
The Principle of BR Analysis
BR analysis is a computational tool that deconvolutes the balance of intermolecular interactions governing a phenomenon described by a QSPR/PLS model [1] [2]. It groups a large number of molecular descriptors (e.g., from VolSurf+) into a small number of easy-to-interpret blocks, such as Size, Polarity, Hydrogen Bond Acidity (HBD), Hydrogen Bond Basicity (HBA), and Lipophilicity [2] [3]. The analysis then graphically shows the relevance of each block to the model, revealing which molecular properties dominate the observed behavior.
Application to Zwitterionic Compounds
For zwitterions, BR analysis can be applied in several critical ways:
Identifying the Best log P Surrogate: BR analysis can identify which chromatographic system (e.g., different HPLC columns or conditions) provides a lipophilicity index that best replicates the balance of forces present in the octanol/water system (log Poct) or in more apolar environments [1]. This ensures that the high-throughput method is a reliable surrogate for the more laborious gold-standard methods.
Understanding Δlog P: BR analysis has demonstrated that the difference between log Poct and log Ptol (Δlog Poct-tol) is predominantly governed by the solute's Hydrogen Bond Donor (HBD) acidity [3]. A high Δlog Poct-tol indicates strong HBD properties, which typically decrease membrane permeability. This descriptor is also useful for estimating the propensity of a molecule to form intramolecular hydrogen bonds (IMHB) [23], a key feature of nonclassical zwitterions that helps stabilize their neutral form and confer high permeability despite low overall lipophilicity [21].
Validating Permeability Methods: BR analysis can compare different permeability assays (e.g., PAMPA vs. cell-based models like Caco-2) to identify which in vitro method best reflects the balance of interactions governing passive permeability in biological systems [1]. This makes the choice of permeability methods safer and aids in drug candidate prioritization.
The following diagram illustrates the workflow for using BR analysis in the lipophilicity assessment of zwitterions.
Diagram 1: BR Analysis Workflow for Zwitterions
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful characterization requires careful selection of materials. The following table details key reagents and their functions.
Table 2: Essential Research Reagents for Lipophilicity Characterization
| Reagent / Material | Function / Purpose | Notes for Zwitterionic Compounds |
|---|---|---|
| 1-Octanol | Lipophilic phase in shake-flask and potentiometry. Mimics biological membranes. | Use high-purity grade. Pre-saturate with aqueous buffer to avoid volume shifts. |
| Toluene | Lipophilic phase for Δlog P measurements. | Used to determine HBD properties and IMHB via Δlog Poct-tol [3]. |
| Buffer Salts (e.g., Phosphates, Citrates, Ammonium Acetate) | Maintain constant pH during experiments. | Choice of buffer ion can influence results via ion-pairing; potassium chloride is often preferred in potentiometry [20]. |
| Potassium Chloride (KCl) | Maintains constant ionic strength in potentiometric titrations. | Minimizes ion-pairing effects compared to other salts [20]. |
| Reversed-Phase HPLC Columns (e.g., C18, LC-ABZ) | Stationary phase for chromatographic lipophilicity indices (CHI, log k'). | LC-ABZ columns are often more versatile for a wide range of polarities [2]. |
| Reference Compounds (e.g., drug cocktail for CHI) | Calibration of chromatographic systems. | Essential for converting retention times into a standardized lipophilicity scale (CHI) [20]. |
Characterizing the lipophilicity of zwitterionic compounds demands a multifaceted approach that goes beyond single-point measurements. A combination of potentiometry and shake-flask methods provides a solid foundation for accurate log D pH-profiling, while chromatographic methods (CHI) offer high-throughput capabilities for screening. The critical insight for modern drug discovery is the emergence of nonclassical zwitterions, which defy traditional pKa-based rules and can achieve high permeability alongside low lipophilicity.
Block Relevance analysis serves as the unifying interpretive framework, transforming raw experimental data into a mechanistic understanding of the intermolecular forces at play. By deconvoluting the contributions of size, polarity, and hydrogen bonding, BR analysis guides the selection of appropriate experimental methods, validates permeability models, and ultimately aids medicinal chemists in rationally designing zwitterionic drug candidates with optimal ADMET properties.
Assessing Passive Permeability Across Cell Types and PAMPA Systems
The ability of a drug candidate to passively permeate through biological membranes is a critical determinant of its absorption and distribution profile. For orally administered drugs, permeability across the gastrointestinal tract (GIT) is essential for systemic exposure, while for central nervous system (CNS)-targeted therapeutics, penetration of the blood-brain barrier (BBB) is paramount [24] [25]. Passive transcellular diffusion, driven by concentration gradients across lipid membranes, represents the primary permeation mechanism for most commercial drugs, estimated to account for 80-95% of absorbed compounds [24] [26].
The Parallel Artificial Membrane Permeability Assay (PAMPA) has emerged as a robust, high-throughput in vitro technique specifically designed to assess passive diffusion potential. First introduced by Kansy in 1998, PAMPA utilizes an artificial membrane structure to simulate passive transport without the complexities of active transport mechanisms present in biological systems [27] [26]. This cell-free approach provides significant advantages in early drug discovery, including cost-effectiveness, high-throughput capability, tolerance to wide pH ranges and DMSO concentrations, and simplified interpretation of results focused solely on passive permeation [24] [27].
The fundamental principle of PAMPA involves measuring compound movement from a donor compartment through a lipid-infused artificial membrane into an acceptor compartment. The rate of permeation is quantified by the effective permeability coefficient (Pe), typically expressed in units of 10−6 cm/s, which allows for ranking compounds based on their intrinsic passive diffusion properties [27] [28]. This core methodology has been adapted to model various biological barriers through modifications to the membrane composition, including GIT-PAMPA for gastrointestinal absorption, BBB-PAMPA for blood-brain barrier penetration, and Skin-PAMPA for transdermal absorption [25] [27].
PAMPA System Design and Configurations
Core PAMPA Architecture and Components
The standard PAMPA system employs a "sandwich" configuration consisting of a donor plate, an artificial membrane, and an acceptor plate. The artificial membrane, supported by a porous hydrophobic filter, is created by impregnating the filter with lipid solutions specifically formulated to mimic the target biological barrier [27]. During incubation, test compounds diffuse from the donor compartment through this membrane into the acceptor compartment, with concentration measurements typically performed using UV spectrophotometry or LC-MS/MS [24] [27].
Commercial PAMPA systems vary in their specific membrane compositions and experimental conditions. The Corning Gentest system features a trilayer lipid-oil-lipid membrane structure that closely mimics the hydrophobic interior and amphiphilic exterior of biological membranes [29]. The Double-Sink PAMPA method patented by pION Inc. incorporates proprietary lipid mixtures and uses Gutbox technology to reduce the unstirred water layer (UWL), enhancing predictability of in vivo permeability [24].
Membrane Composition Variations
The composition of the artificial membrane represents the most significant variable across PAMPA configurations, directly influencing permeability measurements and their biological relevance:
- GIT-PAMPA: Utilizes gastrointestinal tract-optimized lipid mixtures, often based on lecithin or synthetic phospholipids, to predict intestinal absorption [27].
- BBB-PAMPA: Incorporates porcine brain lipid extract dissolved in n-dodecane (typically 2% w/v) to simulate the unique lipid environment of the blood-brain barrier [25] [30].
- Skin-PAMPA: Employs certificates, cholesterol, and stearic acid mixtures to model the complex stratum corneum barrier for transdermal permeability assessment [27].
- Biomimetic PAMPA (BM-PAMPA): Uses highly biomimetic phospholipid mixtures in 1,7-octadiene solutions to replicate in vivo brush-border membrane conditions [26].
- DOPC-PAMPA: Applies highly purified dioleyoylphosphatidylcholine (DOPC) in n-dodecane solution as a standardized membrane composition [26].
Research has demonstrated that the chemical structure of membrane lipids significantly impacts permeability measurements. Studies with synthetic phospholipids containing hydrophobic carbon chains of different lengths (C8, C10, C12) showed that longer hydrophobic chains generally increase drug permeability across all tested compounds [26].
Critical Experimental Parameters
Several experimental factors significantly influence PAMPA permeability measurements and must be carefully controlled for reproducible results:
- pH Conditions: The pH of donor and acceptor compartments dramatically affects permeability for ionizable compounds according to the pH-partition hypothesis. Multiple pH values can be investigated to simulate different gastrointestinal regions [28] [26].
- Incubation Time: Permeability typically increases with time up to a maximum, then may decrease slightly. Standard incubation times range from 1-18 hours depending on the specific PAMPA model [30] [29].
- Unstirred Water Layer (UWL): The aqueous boundary layer adjacent to the membrane surface can limit permeability for highly lipophilic compounds. Stirring using technologies like Gutbox reduces UWL thickness to approximately 60μm [24] [25].
- Temperature: Most PAMPA assays are conducted at room temperature to ensure stability, though some protocols use physiological temperature (37°C) [29] [28].
- Compound Concentration: High concentrations (typically 100-200μM) enable accurate UV detection while avoiding transporter saturation issues irrelevant for passive diffusion [29].
Experimental Protocols
Standard PAMPA Protocol for Permeability Assessment
The following protocol outlines the general procedure for conducting PAMPA experiments, with specific modifications for different membrane models noted where applicable:
Materials and Reagents:
- Test compounds dissolved in DMSO (typically 10 mM stock solutions)
- PAMPA sandwich plates (donor and acceptor compartments)
- Lipid solution appropriate for target barrier (GIT, BBB, or Skin)
- Buffer solutions (commonly phosphate buffer, pH 7.4 or varied for GI simulation)
- UV plate reader or LC-MS/MS system for quantification
- Positive control (e.g., Propranolol for high permeability)
- Negative control (e.g., Atenolol for low permeability)
Procedure:
Membrane Preparation:
- Coat each well of the 96-well filter plate with the appropriate lipid solution
- For BBB-PAMPA: Use porcine brain lipid extract (2% w/v in n-dodecane)
- For GIT-PAMPA: Use proprietary lipid mixtures optimized for intestinal prediction
- Allow membrane to stabilize for approximately 30 minutes
Solution Preparation:
- Dilute test compounds from DMSO stocks to working concentration (typically 50-100 μM) in aqueous buffer
- Maintain final DMSO concentration ≤0.5-5% to avoid membrane disruption
- Filter solutions if necessary to remove undissolved compound
Assembly:
- Add compound solution to donor wells (typically 150-300 μL)
- Fill acceptor wells with blank buffer solution
- Carefully position donor plate onto acceptor plate to form "sandwich"
- Ensure no air bubbles are trapped between compartments
Incubation:
- Incubate assembly at room temperature for specified duration:
- 4-5 hours for standard GIT-PAMPA
- 18 hours for traditional PAMPA-BLM
- 1 hour for PAMPA-BBB-UWL with stirring
- Maintain constant shaking if required by specific protocol
- Use Gutbox technology for assays requiring reduced UWL effects
- Incubate assembly at room temperature for specified duration:
Disassembly and Analysis:
- Separate donor and acceptor plates after incubation
- Measure compound concentrations in both compartments using:
- UV spectrophotometry (high-throughput option)
- LC-MS/MS (increased sensitivity and specificity)
- Quantify initial donor concentration for mass balance calculations
Permeability Calculation:
- Calculate effective permeability (Pe) using the following equation: Where: VD = Volume of donor compartment VA = Volume of acceptor compartment Area = Surface area of membrane × porosity Time = Incubation time [drug]acceptor = Concentration in acceptor compartment [drug]equilibrium = Theoretical equilibrium concentration [28] ```
Data Interpretation:
Protocol Modifications for Specific PAMPA Models
BBB-PAMPA Protocol (based on NCATS methodology):
- Use porcine brain lipid extract in alkane (Pion Inc.)
- Employ 96-well stirwell sandwich plates with coated magnetic stirrers
- Incubate for 60 minutes with continuous stirring using Gutbox technology
- Set UWL thickness to 60μm [25]
High-Throughput Screening Protocol:
- Utilize pre-coated PAMPA plates (e.g., Corning Gentest system)
- Automate liquid handling using 96-well or 384-well formats
- Implement rapid UHPLC-DAD analysis for faster turnaround
- Include quality controls in each plate (positive/negative controls) [29]
Quantitative Permeability Data and Interpretation
Permeability Classification and Reference Compounds
PAMPA permeability values enable quantitative ranking of compounds according to their passive diffusion potential. The following table presents reference permeability data for commonly tested compounds across different PAMPA configurations:
Table 1: Experimental PAMPA Permeability Data for Reference Compounds
| Compound | PAMPA Type | Permeability (10⁻⁶ cm/s) | Classification | Notes |
|---|---|---|---|---|
| Propranolol | GIT-PAMPA | >20 | High | Positive control |
| Atenolol | GIT-PAMPA | <2 | Low | Negative control |
| Verapamil | BBB-PAMPA | 15.8 | High | P-gp substrate |
| Caffeine | BBB-PAMPA | 6.5 | Medium | Mixed transport |
| Carbamazepine | BBB-PAMPA | 12.3 | High | CNS+ compound |
| Saquinavir | GIT-PAMPA | <2 | Low | High efflux |
| Metoprolol | GIT-PAMPA | 15-25 | Medium/High | Borderline permeability [30] [29] |
Classification thresholds: Low permeability: Pe < 1.5-2 × 10⁻⁶ cm/s; Medium permeability: 2-20 × 10⁻⁶ cm/s; High permeability: >20 × 10⁻⁶ cm/s [29] [27] [28].
Comparative Performance of PAMPA Models
Different PAMPA configurations demonstrate varying predictive performance for specific biological barriers:
Table 2: Comparison of PAMPA Model Performance Characteristics
| PAMPA Model | Membrane Composition | Optimal For | Discrimination Accuracy | Key Limitations |
|---|---|---|---|---|
| PAMPA-BBB | Porcine brain lipid extract | BBB permeability | 77% correlation with in vivo B/P ratios | Cannot detect active transport |
| PAMPA-BLM | Traditional black lipid membrane | GI absorption | Identifies BBB+/- compounds | Longer incubation (18h) |
| PAMPA-DS | Gastrointestinal lipid with Double-Sink | GI permeability | Good for rank ordering | May overestimate for some compounds |
| BM-PAMPA | Biomimetic phospholipid mixture | Intestinal prediction | High biomimicry | Complex membrane preparation |
| DOPC-PAMPA | Dioleyoylphosphatidylcholine | Standardized comparison | Reproducible results | Less biomimetic [25] [30] [26] |
The BBB-PAMPA model has demonstrated approximately 77% categorical correlation with in vivo brain-plasma concentration ratios (B/P), validating its utility for predicting brain penetration during early CNS drug discovery [25].
Integration with Block Relevance (BR) Analysis
BR Analysis Framework for PAMPA Data Interpretation
Block Relevance (BR) analysis provides a computational framework for deconvoluting the balance of intermolecular interactions governing permeability phenomena described by QSPR/PLS models. When applied to PAMPA data, BR analysis enables researchers to:
- Identify the most relevant molecular descriptors and physicochemical properties driving passive permeability
- Validate the biomimetic performance of different PAMPA membrane compositions by comparing interaction balances to cell-based systems
- Select optimal chromatographic systems as log Poct surrogates for permeability prediction
- Diagnose the dominant permeability-limiting factors for specific compound classes [1]
The BR approach implemented in MATLAB allows decomposition of QSPR models into blocks representing different intermolecular interaction types (e.g., hydrophobic, hydrogen bonding, ionic), facilitating mechanistic interpretation of permeability data beyond simple statistical correlations.
Correlation with Physicochemical Properties
PAMPA permeability demonstrates strong dependence on fundamental physicochemical properties, though the specific relationships vary across membrane types:
- Lipophilicity: Optimal permeability typically occurs at intermediate log P/log D values (2-4), following a bilinear relationship rather than simple linear correlation [31]
- Hydrogen Bonding: Both hydrogen bond donor (HBD) and acceptor (HBA) capacity inversely correlate with permeability, with typical QSPR models incorporating SAHA and SAHD descriptors [24]
- Molecular Size: Larger molecules generally show reduced permeability, though this relationship is modified by lipophilicity and hydrogen bonding capacity
- Ionization State: Permeability follows pH-partition behavior, with uncharged species exhibiting significantly higher permeability than ionized forms [24] [26]
The original PAMPA QSPR model developed by Akamatsu's group illustrates these relationships:
Where logPoct represents octanol-water partitioning, pKa is the acid dissociation constant, and SAHA/SAHD represent hydrogen bond acceptor/donor surface areas [24].
Research Reagent Solutions
Table 3: Essential Research Reagents for PAMPA Experiments
| Reagent/Equipment | Function/Application | Example Sources |
|---|---|---|
| Porcine Brain Lipid Extract | BBB-PAMPA membrane formation | Pion Inc. (Catalog #110672) |
| 96-well Stirwell Sandwich Plates | PAMPA assay platform with stirring capability | Pion Inc. (Catalog #110243) |
| Pre-coated PAMPA Plates | Ready-to-use artificial membranes | Corning Gentest |
| Brain Sink Buffer | Acceptor solution for BBB-PAMPA | Pion Inc. (Catalog #110674) |
| Gutbox Technology | Reduces unstirred water layer effects | Pion Inc. |
| LC-MS/MS Systems | Sensitive quantification of permeability | Various vendors |
| UV Plate Readers | High-throughput concentration measurement | Tecan Infinite 200 PRO |
PAMPA represents a versatile, high-throughput platform for assessing passive permeability across various biological barriers. Through strategic selection of membrane composition and experimental conditions, researchers can obtain valuable early-stage permeability data to guide compound optimization. The integration of PAMPA results with Block Relevance analysis provides enhanced mechanistic understanding of the intermolecular interactions governing passive diffusion, enabling more informed decisions in drug discovery campaigns.
When implemented as part of a comprehensive ADME screening strategy, with complementary cell-based assays for evaluating active transport mechanisms, PAMPA significantly enhances the efficiency of lead optimization and candidate selection processes. The continued refinement of PAMPA models and their correlation with in vivo permeability outcomes remains an active area of research in pharmaceutical sciences.
Determining Lipophilicity in Nonpolar Membrane-like Environments
Lipophilicity, a fundamental physicochemical property in drug discovery, is traditionally characterized by the logarithmic partition coefficient (log P) in the octanol/water system [32]. However, the internal core of cell membranes is a largely hydrophobic and non-polar environment [33]. Relying solely on traditional lipophilicity measurements can therefore be misleading, as a compound's behavior in a nonpolar milieu can differ significantly from its behavior in octanol/water [33]. Block Relevance (BR) analysis is a computational tool that deconvolutes the balance of intermolecular interactions governing a drug discovery-related phenomenon described by a QSPR/PLS model [1]. It aids in identifying the most appropriate experimental methods for measuring lipophilicity in apolar environments, thereby making drug candidate prioritization safer and more efficient [1]. These Application Notes provide detailed protocols for determining lipophilicity in nonpolar, membrane-mimetic conditions, framed within the strategic context of BR analysis.
Key Experimental Methodologies
Shake-Flask Method in Toluene/Water System
Principle: This method directly measures the partition coefficient of a compound between toluene (a nonpolar organic phase) and an aqueous buffer phase [33]. Toluene more closely mimics the inert hydrocarbon core of a lipid bilayer compared to the hydrogen-bonding octanol phase.
Protocol:
- Preparation: Pre-saturate high-purity toluene and an aqueous buffer (e.g., phosphate buffer pH 7.4) with each other by mixing overnight in a separatory funnel. Allow phases to separate completely before use.
- Partitioning: In a sealed vial (e.g., 4 mL), combine 1 mL of the toluene phase and 1 mL of the aqueous phase. Add a precise amount of the test compound, either as a solid or from a stock solution.
- Equilibration: Agitate the mixture vigorously on a mechanical shaker for a predetermined time (e.g., 1-2 hours) at a constant temperature (e.g., 25°C). Subsequently, centrifuge the vials to achieve complete phase separation.
- Quantification: Carefully separate the two phases. Determine the concentration of the compound in each phase using a suitable analytical method, such as UV-Vis spectroscopy or HPLC.
- Calculation: Calculate the log Ptol using the formula: log Ptol = log (Ctol / Caq), where Ctol and Caq are the equilibrium concentrations in the toluene and aqueous phases, respectively.
Chromatographic Determination of a Lipophilicity Index (log k'80PLRP-S)
Principle: Reverse-phase chromatography using a polystyrene-divinylbenzene polymeric column (PLRP-S) provides a lipophilicity index that correlates with partitioning into apolar environments [33]. The polymeric stationary phase lacks the silanol groups of silica-based columns, offering a more inert and reproducible nonpolar surface.
Protocol:
- Chromatographic System: Utilize a PLRP-S column (e.g., 150 x 4.6 mm, 5 µm particle size). The mobile phase should be a binary mixture of acetonitrile and a pH-buffered aqueous solution.
- Mobile Phase Preparation: Prepare a high-content organic mobile phase, such as 80% acetonitrile and 20% water (v/v), often termed "log k'80". The aqueous portion may contain a buffer like ammonium acetate to control ionization.
- Analysis: Inject the test compound dissolved in a suitable solvent. Measure the retention time (tR). Also, measure the void time (t0) using an unretained compound like uracil or sodium nitrate.
- Calculation: Calculate the capacity factor, log k', for each compound: k' = (tR - t0) / t0. The logarithm of this value, log k'80, serves as the chromatographic lipophilicity index.
Reverse-Phase Thin-Layer Chromatography (RP-TLC)
Principle: RP-TLC is a simple, high-throughput technique for determining experimental lipophilicity parameters [32] [34]. Compounds are separated on TLC plates with a nonpolar stationary phase, and their migration is related to lipophilicity.
Protocol:
- Stationary Phase: Use RP-TLC plates, such as RP-18F254, RP-8F254, or RP-2F254.
- Mobile Phase: Apply mobile phases consisting of acetone, acetonitrile, or 1,4-dioxane as organic modifiers mixed with water or a buffer.
- Development: Spot test compounds on the baseline of the TLC plate and develop the chamber using a chosen mobile phase.
- Measurement and Calculation: After development and drying, measure the retention factor (RF). Calculate the RM value using the formula: RM = log (1/RF - 1). The RM0 value, obtained by extrapolating RM values to 0% organic modifier, is often used as an experimental log P surrogate.
The following workflow integrates these methodologies within a BR analysis framework to guide the selection of the most relevant lipophilicity measurement.
Quantitative Data and Comparative Analysis
Lipophilicity can vary significantly depending on the measurement environment, as demonstrated by studies on pharmaceutical compounds [33]. The table below summarizes hypothetical lipophilicity data for different compound classes across various systems, illustrating how the choice of method impacts the resulting descriptor.
Table 1: Comparison of Lipophilicity Values for Model Compounds in Different Systems
| Compound Class | Example | log Poct (Shake-Flask) | log Ptol (Shake-Flask) | log k'80 (PLRP-S) | RM0 (RP-TLC) |
|---|---|---|---|---|---|
| Acidic | Ibuprofen | 3.5 - 4.0 | ~2.0 - 2.5 | ~2.8 - 3.3 | ~2.5 - 3.0 |
| Basic | Propranolol | 3.0 - 3.5 | ~1.5 - 2.0 | ~2.2 - 2.7 | ~2.0 - 2.5 |
| Neutral (Lipophilic) | - | 4.5 - 5.0 | ~4.0 - 4.5 | ~4.2 - 4.7 | ~4.0 - 4.5 |
| Neutral (Polar) | - | 1.0 - 1.5 | ~0.5 - 1.0 | ~0.8 - 1.3 | ~0.6 - 1.1 |
Interpretation: The data consistently shows that log P in the toluene/water system (log Ptol) is generally lower than log P in the octanol/water system (log Poct). This difference arises because toluene, being an inert hydrocarbon, cannot stabilize charged or polar groups via hydrogen bonding, unlike octanol [33]. BR analysis helps interpret these differences by deconvoluting the specific intermolecular interactions (e.g., hydrogen bonding, electrostatic) responsible for the observed partition coefficients [1].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Lipophilicity Assessment in Nonpolar Media
| Reagent / Material | Function and Rationale |
|---|---|
| Toluene (ACS grade) | Serves as the nonpolar organic phase in shake-flask experiments, mimicking the inert hydrocarbon interior of cell membranes [33]. |
| PLRP-S Column | A polystyrene/divinylbenzene polymeric stationary phase for HPLC. It provides an inert, nonpolar surface for determining chromatographic lipophilicity indices relevant to membrane core partitioning [33]. |
| RP-18F254 TLC Plates | Reverse-phase TLC plates with a C18-modified silica stationary phase. Used for high-throughput experimental estimation of lipophilicity (RM0) [32] [34]. |
| Acetonitrile (HPLC grade) | A common organic modifier for chromatographic mobile phases (HPLC, TLC) and a solvent for measuring pKa in pure non-aqueous environments [33]. |
| n-Octanol (ACS grade) | The standard organic solvent for the traditional shake-flask method, providing a reference point for lipophilicity (log Poct) [33] [35]. |
| Buffer Salts | To prepare aqueous phases at physiologically relevant pH values, controlling the ionization state of ionizable compounds during partitioning experiments [35]. |
Integrating lipophilicity measurements in nonpolar, membrane-mimetic environments is crucial for a accurate understanding of a drug candidate's behavior. The experimental strategies outlined—shake-flask in toluene/water, chromatography on PLRP-S columns, and RP-TLC—provide essential tools for this purpose. The application of Block Relevance analysis enhances this process by providing a rational, data-driven framework for selecting the most appropriate method based on the molecular interactions of the compound under investigation [1]. This integrated approach allows for a more reliable prediction of membrane permeability and ultimately speeds up the prioritization of successful drug candidates.
Estimating Hydrogen Bond Donor Properties via Δlog Poct-tol
Within modern drug discovery, the rational design of compounds with favorable absorption, distribution, metabolism, and excretion (ADME) properties is paramount. Lipophilicity, commonly expressed as the logarithm of the octanol/water partition coefficient (log Pₒcₜ), is a critical parameter in these efforts, famously forming part of the Lipinski's Rule of Five [36]. However, the traditional log Pₒcₜ provides a generalized picture of lipophilicity, often obscuring the nuanced roles of specific intermolecular forces, particularly a compound's capacity to act as a hydrogen bond donor (HBD).
The Block Relevance (BR) analysis is a computational methodology that deconvolutes the balance of intermolecular interactions governing a drug discovery-related phenomenon described by a QSPR/PLS model [1]. It groups molecular descriptors into chemically intuitive blocks (e.g., Size, HBD, HBA), allowing researchers to visually and quantitatively interpret which interaction forces dominate a given process [2] [37]. This application note, framed within a broader thesis on BR analysis for lipophilicity assessment, details how the difference between lipophilicity in octanol/water and toluene/water systems—Δlog Pₒcₜ-ₜₒₗ—serves as a powerful, experimentally accessible proxy for estimating hydrogen bond donor properties, thereby enabling more informed candidate optimization.
Theoretical Foundation
The Limitation of log Pₒcₜ and the Rise of Δlog Pₒcₜ-ₜₒₗ
While log Pₒcₜ is an established tool, its value encodes a composite of multiple intermolecular interactions, with a significant contribution from molecular size [36]. BR analysis has theoretically and practically demonstrated that the "Size" contribution heavily dominates the log Pₒcₜ value, thereby masking the specific signal from a compound's hydrogen bond acidity (i.e., its HBD capacity) [36] [37].
To overcome this limitation, the parameter Δlog Pₒcₜ-ₜₒₗ is introduced. It is defined as the difference between the partition coefficient in the octanol/water system and the partition coefficient in the toluene/water system: Δlog Pₒcₜ-ₜₒₗ = log Pₒcₜ - log Pₜₒₗ
Toluene is a significantly more apolar solvent than octanol. It cannot act as a hydrogen bond donor and is a very poor hydrogen bond acceptor. Consequently, the toluene/water system is highly sensitive to a solute's ability to donate hydrogen bonds, as these HBD interactions cannot be effectively compensated by the solvent. BR analysis applied to a large dataset confirmed that the hydrogen bond donor (HBD) properties of the solutes mainly govern Δlog Pₒcₜ-ₜₒₗ [37]. By subtracting log Pₜₒₗ from log Pₒcₜ, the pervasive influence of molecular size is effectively canceled out, isolating the HBD component of lipophilicity.
Link to Intramolecular Hydrogen Bonding (IMHB)
Beyond quantifying intermolecular HBD strength, Δlog Pₒcₜ-ₜₒₗ is a crucial tool for identifying compounds capable of forming intramolecular hydrogen bonds (IMHB) [36]. When a molecule forms a stable IMHB, its hydrogen bond donor group is effectively "shielded" and less available to interact with the aqueous or organic phase. This results in a higher measured log Pₜₒₗ (as the molecule behaves as if it is less polar) and consequently, a lower Δlog Pₒcₜ-ₜₒₗ value. Therefore, a low or negative Δlog Pₒcₜ-ₜₒₗ can be a strong indicator of IMHB formation, a property that can significantly improve membrane permeability and oral bioavailability [36].
Table 1: Interpretation of Δlog Pₒcₜ-ₜₒₗ Values
| Δlog Pₒcₜ-ₜₒₗ Value | Chemical Interpretation | Potential Implications for Drug Properties |
|---|---|---|
| High Positive Value | Strong intermolecular hydrogen bond donor (HBD) capacity. | Potentially lower membrane permeability, higher aqueous solubility. |
| Low or Negative Value | Weak intermolecular HBD; potential formation of intramolecular H-bonds (IMHB). | Improved membrane permeability, potential for oral bioavailability. |
Quantitative Data and Methodology
Key Experimental and Calculated Values
The following table summarizes the core quantitative data and relationships essential for applying this methodology.
Table 2: Key Partition Systems and Descriptor Correlations
| Parameter | Description | Typical Value Range / Equation | Primary Information Content |
|---|---|---|---|
| log Pₒcₜ | Logarithm of the octanol/water partition coefficient. | Foundational QSAR parameter: log Po/w = 0.09 + 0.56E - 1.05S + 0.03A - 3.46B + 3.81V [38]. |
Composite of size, polarity, and H-bond basicity; HBD (A) contribution is minimal. |
| log Pₜₒₗ | Logarithm of the toluene/water partition coefficient. | Experimentally determined via shake-flask or chromatographic methods. | Highly sensitive to solute H-bond donor (HBD) capacity. |
| Δlog Pₒcₜ-ₜₒₗ | Difference between the two partition coefficients. | Δlog Pₒcₜ-ₜₒₗ = log Pₒcₜ - log Pₜₒₗ [36] [37]. |
Isolated measure of solute hydrogen bond acidity (HBD). Low values suggest IMHB. |
| Abraham's A Descriptor | Calculated solute hydrogen bond acidity. | Used in LFER models: log SP = c + eE + sS + aA + bB + vV [38]. |
Theoretical 2D descriptor for HBD capacity; can be used to model Δlog P. |
Experimental Protocol: Determining Δlog Pₒcₜ-ₜₒₗ
This protocol outlines the steps for the experimental determination of Δlog Pₒcₜ-ₜₒₗ using the shake-flask method, followed by data interpretation.
Step 1: Reagent and Solution Preparation
- Prepare a phosphate buffer (e.g., 0.1 M, pH 7.4) to simulate physiological conditions.
- Saturate high-purity 1-octanol and toluene with the aqueous buffer by mixing them in a separatory funnel for 24 hours. Allow phases to separate completely before use.
- Prepare a stock solution of the compound of interest in a volatile, water-miscible solvent (e.g., DMSO, methanol), ensuring the final concentration in the partitioning experiment is sufficiently below its solubility limit in both phases.
Step 2: Partitioning Experiment
- For each solvent system (octanol/buffer and toluene/buffer), add precisely measured volumes of the organic phase and aqueous buffer (e.g., 1.5 mL each) to a glass vial.
- Spike a known, small volume of the compound stock solution into each vial.
- Seal the vials and agitate them vigorously on a mechanical shaker for 2-4 hours at a constant temperature (e.g., 25°C) to reach partitioning equilibrium.
- Centrifuge the vials to achieve complete phase separation.
Step 3: Analytical Quantification
- Carefully separate the two phases for each system.
- Quantify the concentration of the compound in each phase using a suitable analytical method, typically reverse-phase HPLC with UV detection. The method should be calibrated with standard solutions of known concentration.
- Calculate log P for each system using the formula:
log P = log (Cₒᵣgₐₙᵢc / Cₐqᵤₑₒᵤₛ).
Step 4: Data Analysis and Interpretation
- Calculate Δlog Pₒcₜ-ₜₒₗ: Subtract the calculated log Pₜₒₗ from log Pₒcₜ.
- Interpret the result using the guidance in Table 1. A high positive value indicates strong HBD character, while a low or negative value suggests potential IMHB formation. For conclusive IMHB assessment, compare Δlog Pₒcₜ-ₜₒₗ values of a target compound with a structurally similar control compound incapable of forming the suspected IMHB [36].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Computational Tools
| Item / Reagent | Function / Role in the Protocol |
|---|---|
| 1-Octanol (water-saturated) | The standard organic solvent for lipophilicity measurement. A proton-acceptor solvent that mimics some biological environments. |
| Toluene (water-saturated) | The apolar, non-HBD complementary solvent. Its high sensitivity to solute HBD is key to calculating Δlog Pₒcₜ-ₜₒₗ. |
| Aqueous Buffer (e.g., Phosphate, pH 7.4) | Represents the aqueous physiological environment. Ensures consistent pH, critical for ionizable compounds. |
| HPLC-UV System | The primary analytical instrument for precise quantification of solute concentration in each phase after partitioning. |
| VolSurf+ Software | Used to compute 3D molecular descriptors (e.g., DRDODO, WO1) related to HBD properties, which can be correlated with Δlog Pₒcₜ-ₜₒₗ [37] [38]. |
| BR Analysis (MATLAB implementation) | The computational tool for deconvoluting the PLS models built from experimental data, visually revealing the relevance of the HBD block [1] [2]. |
Integration with Block Relevance Analysis
The true power of this methodology is fully realized when integrated with BR analysis. The workflow below illustrates how experimental data is processed and interpreted through the BR framework to guide medicinal chemistry decisions.
The BR analysis takes the PLS model built from experimental Δlog Pₒcₜ-ₜₒₗ data and molecular descriptors and groups the descriptors into chemically meaningful blocks (e.g., Size, Polarizability, HBD, HBA) [2]. The output visually demonstrates the high relevance of the HBD block in explaining the variance in Δlog Pₒcₜ-ₜₒₗ, thereby providing unambiguous theoretical support for its use as an HBD-specific metric [37]. This integrated approach does not just provide a number; it offers a deep, mechanistically grounded understanding of the intermolecular forces at play, making the choice of methods for measuring permeability safer and speeding up the prioritization of drug candidates [1].
Troubleshooting Complex Molecular Properties with BR Analysis
In modern drug discovery, lipophilicity is a fundamental physicochemical property that profoundly influences a compound's absorption, distribution, metabolism, and excretion (ADME). While the octanol/water partition coefficient (log Pₒcₜ) has served as the traditional benchmark for lipophilicity, its limitations in capturing the complexity of molecular interactions in biological systems have spurred the development of more informative metrics [39]. Among these, Δlog Pₒcₜ₋ₜₒₗ—the difference between a compound's partition coefficient in octanol/water and toluene/water systems—has emerged as a crucial parameter for assessing key molecular properties.
This Application Note details how Block Relevance (BR) analysis, a sophisticated computational tool for deconvoluting the balance of intermolecular interactions in Quantitative Structure-Property Relationship (QSPR) models, was employed to definitively identify hydrogen bond donor (HBD) capacity as the dominant molecular force governing Δlog Pₒcₜ₋ₜₒₗ values. This finding provides medicinal chemists with a powerful, interpretable framework for optimizing compound properties during early drug development stages.
Theoretical Background
The Significance of Δlog Pₒcₜ₋ₜₒₗ
The Δlog Pₒcₜ₋ₜₒₗ parameter provides unique information about a compound's intermolecular interaction potential that simple log P values cannot capture:
- Probe for Hydrogen Bonding: Toluene is a much weaker hydrogen bond acceptor than octanol, making the toluene/water system particularly sensitive to a solute's hydrogen bond donor capacity [37].
- Intramolecular Hydrogen Bond (IMHB) Assessment: Δlog Pₒcₜ₋ₜₒₗ serves as an experimental tool for identifying compounds capable of forming intramolecular hydrogen bonds, which significantly impact membrane permeability and bioavailability [37].
- Beyond Rule of 5 (bRo5) Applications: For larger, more complex molecules beyond traditional drug-like space, Δlog P parameters provide critical insights into permeability challenges and chameleonic properties that enable oral bioavailability [39].
Block Relevance Analysis Fundamentals
Block Relevance (BR) analysis is a computational method implemented in MATLAB that extends traditional Partial Least Squares (PLS) modeling in QSPR studies [1]. Its core functionality includes:
- Interaction Deconvolution: BR analysis decomposes the complex balance of intermolecular interactions governing drug discovery-related phenomena described by QSPR/PLS models [1].
- Descriptor Grouping: Molecular descriptors are grouped into chemically meaningful "blocks" representing specific types of intermolecular forces or physicochemical properties [37].
- Visualization of Dominant Effects: The analysis generates intuitive graphical representations showing the relative contribution of each descriptor block to the overall property being modeled [2].
Experimental Protocol
Dataset Curation and Preprocessing
The foundational study applied a rigorous multi-step process to ensure data quality and interpretability [37]:
Step 1: Literature Data Collection
- Collected >200 experimental log Pₜₒₗ values with corresponding log Pₒcₜ values from published literature
- Compiled structures and associated partition coefficients into a standardized database
Step 2: IMHB Compound Removal
- Applied in-house software to identify and remove molecules capable of forming intramolecular hydrogen bonds
- This critical step isolated the effect of intermolecular hydrogen bonding on Δlog Pₒcₜ₋ₜₒₗ
Step 3: Δlog P Calculation
- Computed Δlog Pₒcₜ₋ₜₒₗ values using the formula: Δlog Pₒcₜ₋ₜₒₗ = log Pₒcₜ - log Pₜₒₗ
Table 1: Dataset Characteristics After Curation
| Parameter | Description | Value/Count |
|---|---|---|
| Initial Compounds | Collected from literature | >200 molecules |
| Final Dataset | After IMHB filtering | Not specified in study |
| Descriptors | VolSurf+ descriptors evaluated | 82 total descriptors |
| Block Categories | Descriptor groupings in BR analysis | 6 chemically meaningful blocks |
Computational Modeling and BR Analysis Workflow
Step 4: PLS Model Development
- Correlated calculated Δlog Pₒcₜ₋ₜₒₗ values with 82 VolSurf+ descriptors using PLS regression
- Validated model performance using standard QSPR validation techniques
Step 5: Block Relevance Analysis Implementation
- Grouped the 82 VolSurf+ descriptors into six chemically interpretable blocks using BR analysis
- Generated visualizations showing the relative relevance of each descriptor block to the PLS model
- Interpreted the dominant physicochemical forces driving Δlog Pₒcₜ₋ₜₒₗ values
Diagram 1: BR Analysis Workflow for Δlog Pₒcₜ₋ₜₒₗ Analysis. This flowchart illustrates the sequential steps from data collection through Block Relevance analysis to identify hydrogen bond donation as the dominant molecular force.
Results and Interpretation
BR Analysis Reveals HBD Dominance
The BR analysis provided clear, quantitative evidence of the predominant role played by hydrogen bond donor capacity:
- Dominant Block Identification: The descriptor block representing hydrogen bond donor (HBD) properties showed significantly higher relevance than other blocks in the PLS model for Δlog Pₒcₜ₋ₜₒₗ [37].
- Visual Confirmation: Graphical outputs from the BR analysis intuitively displayed the overwhelming contribution of HBD properties compared to other molecular descriptors [37].
- Mechanistic Explanation: The physical basis for this dominance lies in the differential hydrogen bond accepting capabilities of the two solvent systems—octanol is a strong hydrogen bond acceptor while toluene is a very weak one [37].
Table 2: Key Findings from BR Analysis of Δlog Pₒcₜ₋ₜₒₗ
| Analysis Component | Finding | Interpretation |
|---|---|---|
| Dominant Descriptor Block | Hydrogen Bond Donor (HBD) Properties | HBD capacity is the primary driver of Δlog Pₒcₜ₋ₜₒₗ |
| Supporting Evidence | Visualization of block relevance | Graphical representation shows clear dominance of HBD block |
| Physical Basis | Differential HB accepting of solvents | Octanol (strong acceptor) vs. toluene (weak acceptor) |
| Practical Application | Estimation of solute HBD properties | Δlog Pₒcₜ₋ₜₒₗ serves as experimental measure of HBD capacity |
Implications for Intramolecular Hydrogen Bonding
The study further demonstrated how this finding supports the interpretation of intramolecular hydrogen bonding (IMHB):
- Compounds capable of forming strong IMHB show attenuated Δlog Pₒcₜ₋ₜₒₗ values because their hydrogen bond donor groups are already engaged internally [37].
- This provides experimental validation for IMHB predictions and helps explain the permeability advantages of certain bRo5 compounds that exhibit chameleonic behavior [39].
Research Reagent Solutions
Table 3: Essential Research Tools for BR Analysis and Lipophilicity Assessment
| Tool/Category | Specific Examples | Function/Application |
|---|---|---|
| Computational Tools | MATLAB with BR analysis implementation | Deconvolution of intermolecular interactions in QSPR models [1] |
| Molecular Descriptors | VolSurf+ descriptors (82 total) | Quantification of structural and physicochemical properties [37] |
| Chromatographic Systems | Supelcosil LC-ABZ column | Reliable measurement of lipophilicity indices [2] |
| Partition Systems | Octanol/water, Toluene/water | Standardized systems for log P measurements [37] |
| Data Analysis | PLS Regression, QSPR Modeling | Correlation of structural features with physicochemical properties [37] |
Application in Drug Discovery
The identification of HBD capacity as the dominant force in Δlog Pₒcₜ₋ₜₒₗ has significant practical applications in drug discovery:
Diagram 2: Drug Discovery Applications of BR Analysis Findings. The identification of HBD as the dominant driver of Δlog Pₒcₜ₋ₜₒₗ enables multiple practical applications in molecular design and optimization.
Strategic Compound Design and Optimization
- Hydrogen Bond Management: Medicinal chemists can use Δlog Pₒcₜ₋ₜₒₗ measurements to strategically manage hydrogen bond donor counts in molecular design, particularly for membrane permeability optimization [39].
- bRo5 Space Navigation: For compounds beyond Rule of 5 space (bRo5), including PROTACs, macrocycles, and other new chemical modalities, understanding HBD impact on lipophilicity and permeability is essential for achieving oral bioavailability [39].
- Candidate Prioritization: BR analysis speeds up drug candidate prioritization by providing clearer insight into the balance of intermolecular interactions governing permeability and other ADME properties [1].
Block Relevance analysis has definitively established hydrogen bond donor capacity as the dominant molecular force driving Δlog Pₒcₜ₋ₜₒₗ values. This fundamental insight provides medicinal chemists with a powerful, interpretable framework for understanding and optimizing key physicochemical properties during drug discovery. The integration of BR analysis into lipophilicity assessment protocols enables more efficient candidate prioritization and strategic compound design, particularly in challenging beyond-Rule-of-5 chemical space where traditional guidelines fail. As drug discovery increasingly ventures into more complex chemical modalities, the ability to deconvolute and understand the specific intermolecular forces governing physicochemical behavior becomes increasingly valuable for designing successful therapeutic agents.
Strategies for Beyond-Rule-of-5 (bRo5) Compounds and Intramolecular Hydrogen Bonding (IMHB)
The pursuit of compounds beyond the Rule of 5 (bRo5) has become an essential strategy in modern drug discovery, particularly for addressing challenging biological targets. Lipinski's Rule of 5 (Ro5) was established to identify compounds with a higher probability of oral absorption, defining thresholds for molecular weight (MW < 500 Da), hydrogen bond donors (HBD < 5), hydrogen bond acceptors (HBA < 10), and calculated lipophilicity (cLogP < 5) [40] [41]. However, strict adherence to the Ro5 would eliminate opportunities for numerous valuable therapeutics, as oral druggable space extends far beyond these boundaries [41].
The bRo5 chemical space typically encompasses compounds with a molecular weight of 500-1000 Da, up to 6 hydrogen bond donors, up to 15 hydrogen bond acceptors, and a cLogP between -2 and +10 [42]. This expanded space is particularly relevant for targeting protein-protein interactions (PPIs), kinases, and other complex biological targets with large, shallow, or flexible binding sites that are difficult to address with conventional small molecules [40] [42]. Analysis of 37 target proteins with bRo5 drugs reveals that targets with "complex" hot spot structures (four or more hot spots) particularly benefit from bRo5 compounds, enabling improved affinity or selectivity compared to smaller molecules [40].
Strategic Importance of bRo5 Compounds in Drug Discovery
Applications for Challenging Target Classes
bRo5 compounds provide unique advantages for addressing target classes that have historically been considered "undruggable":
Protein-Protein Interaction Inhibitors: PPIs often involve large, flat binding surfaces spanning 1,500-3,000 Ų, requiring larger compounds to achieve sufficient binding affinity. Approximately 50% of PPI inhibitors in the scientific literature are bRo5 compounds [40] [42]. The human proteome includes an estimated ~1,000,000 different PPIs, making this one of the most important sources of novel targets for future drug discovery [42].
Kinase Inhibitors: Over 30% of approved kinase inhibitors are bRo5 compounds, where increased size often enhances selectivity by accessing unique binding regions beyond the ATP pocket [40] [43].
Targets with Complex Hot Spot Structures: Analysis of bRo5 targets reveals that 24 out of 37 proteins have complex hot spot structures (mean of 5.63 hot spots), which benefit from larger compounds that can engage multiple hot spots simultaneously [40].
Property-Based Classification of bRo5 Targets
Targets benefiting from bRo5 compounds can be classified based on their hot spot characteristics and ligand binding profiles:
Table 1: Classification of bRo5 Targets Based on Hot Spot Structure
| Target Class | Hot Spot Characteristics | Correlation Between Affinity & MW | Primary Motivation for bRo5 Compounds | Example Targets |
|---|---|---|---|---|
| Complex I | 4+ hot spots, including strong ones | Positive correlation | Improved pharmaceutical properties, enhanced affinity | HIV-1 Protease, Heat Shock Protein 90 |
| Complex II | Strong hot spots but different spatial arrangement | No correlation | Increased selectivity, engaging unique binding regions | Protein Kinases |
| Complex III | Variable, target-specific features | Target-dependent | Address specific binding challenges | Target-specific cases |
| Simple | 3 or fewer weak hot spots | Requires larger compounds | Achieve acceptable affinity by interacting beyond hot spot region | Targets with shallow binding sites |
The following diagram illustrates the decision framework for pursuing bRo5 strategies based on target properties:
Intramolecular Hydrogen Bonding in bRo5 Compounds
Role of IMHB in bRo5 Compound Properties
Intramolecular hydrogen bonding (IMHB) represents a critical structural feature that significantly influences the physicochemical properties, conformation, and bioavailability of bRo5 compounds. IMHB occurs when a hydrogen atom covalently bound to an electronegative donor (typically N or O) interacts with an acceptor atom (typically O, S, or N) within the same molecule [43].
In the bRo5 chemical space, IMHB serves several essential functions:
Conformational Stabilization: IMHB can stabilize specific molecular conformations, including those that mimic secondary structure motifs (α-helices, β-strands, reverse turns) found in biological macromolecules, enabling inhibition of challenging targets like PPIs [42].
Modulation of Molecular Properties: By reducing the effective polar surface area and shielding polarity, IMHB enhances membrane permeability and oral bioavailability despite high molecular weight and numerous hydrogen bonding groups [41] [42].
Enabling Molecular Chameleonicity: bRo5 compounds with IMHB capability can adopt different conformations in various environments (e.g., polar vs. apolar), displaying environment-dependent conformational polymorphism that is crucial for balancing solubility and permeability requirements [42].
Experimental and Computational Assessment of IMHB
Table 2: Methods for Characterizing Intramolecular Hydrogen Bonding
| Method Category | Specific Techniques | Information Provided | Applications in bRo5 Space |
|---|---|---|---|
| Spectroscopic Methods | NMR (TBDIE on ¹³C shifts), IR, Raman, IINS, NQR | Hydrogen bond strength, proton position, bond characteristics | Assessing IMHB strength in solution, identifying RAHB systems [43] |
| Computational Approaches | Molecular Tailoring Approach, QTAIM, NCI, Local Mode Analysis | Individual IMHB energy estimation, interaction characterization | Deconvoluting multiple IMHBs in complex molecules, ranking by strength [43] |
| Advanced Theoretical Methods | Car-Parrinello Molecular Dynamics, ab initio G4 theory | Proton transfer phenomena, nuclear quantum effects, substituent effects | Studying excited-state intramolecular proton transfer (ESIPT) [43] |
| Thermal Analysis | Differential Scanning Calorimetry (DSC) | Influence of IMHB on polymorphic states | Understanding solid-state behavior and formulation properties [43] |
Experimental Protocols for bRo5 Compound Assessment
Protocol 1: Lipophilicity Assessment Using Block Relevance Analysis
Objective: Determine the lipophilicity of bRo5 compounds and interpret the balance of intermolecular forces using Block Relevance (BR) analysis.
Background: Conventional log P measurements may not accurately reflect the partitioning behavior of bRo5 compounds due to their complex molecular features. BR analysis deconvolutes the balance of intermolecular interactions governing partitioning phenomena, providing medicinal chemists with a practical tool for interpreting lipophilicity in the context of drug discovery [1] [2].
Materials:
- Analytical System: HPLC system with appropriate detection (UV/Vis or MS)
- Chromatographic Columns: Supelcosil LC-ABZ or equivalent stationary phase
- Mobile Phase: Buffered aqueous component and organic modifier (typically acetonitrile or methanol)
- Reference Compounds: Set of standards with known lipophilicity values
- Software: MATLAB with BR analysis implementation [1]
Procedure:
- Method Development:
- Establish gradient elution method covering 0-100% organic modifier
- Ensure adequate retention (k > 0.5) for all analytes
- Maintain column temperature at 25°C ± 1°C
System Calibration:
- Inject reference compounds with known lipophilicity values
- Determine retention times and calculate log k values
- Establish correlation between log k and reference log P values
Sample Analysis:
- Prepare analyte solutions in appropriate solvent (typically DMSO)
- Inject samples in triplicate to ensure reproducibility
- Record retention times and calculate chromatographic indices
BR Analysis Implementation:
- Input chromatographic data into MATLAB BR analysis tool
- Deconvolute intermolecular solute-system interactions
- Identify the chromatographic system that provides reliable log P octanol/water surrogates
- Interpret the balance of intermolecular forces governing partitioning
Data Interpretation:
- BR analysis identifies which chromatographic system best mimics biological partitioning
- The method determines if different lipophilicity measurement systems express the same balance of intermolecular forces
- Results guide selection of appropriate lipophilicity assessment methods for specific bRo5 compound classes [1] [2]
Protocol 2: Permeability Assessment with BR Analysis
Objective: Evaluate passive permeability of bRo5 compounds and validate methods using Block Relevance analysis.
Background: Permeability assessment is crucial for bRo5 compounds where traditional prediction models often fail. BR analysis enables researchers to check the universality of passive permeability measurements across different cell types and identify PAMPA methods that provide the same balance of intermolecular interactions as cell-based systems [1].
Materials:
- Cell-Based Systems: Caco-2, MDCK, or other relevant cell lines
- PAMPA Plates: Commercial parallel artificial membrane permeability assay systems
- Buffers: Transport buffers (pH 7.4), recovery solutions
- Analytical Instrumentation: LC-MS/MS for compound quantification
- Software: BR analysis implementation for permeability data interpretation
Procedure:
- System Selection and Validation:
- Select appropriate cell-based or PAMPA system for compound class
- Validate system with reference compounds of known permeability
- Establish linearity of transport rate measurements
Permeability Assay:
- Prepare compound solutions at relevant concentrations (typically 1-10 μM)
- Conduct permeability experiments in both apical-to-basolateral and basolateral-to-apical directions
- Sample at multiple time points (e.g., 30, 60, 90, 120 minutes)
- Analyze samples using validated LC-MS/MS methods
Data Analysis:
- Calculate apparent permeability (Papp) coefficients
- Determine efflux ratios for relevant compounds
- Assess mass balance and compound recovery
BR Analysis Application:
- Input permeability data from multiple systems into BR analysis
- Identify the PAMPA method that provides the same picture in terms of balance of intermolecular interactions as cell-based systems
- Validate the universality of passive permeability measurements among different cell types
Data Interpretation:
- BR analysis confirms whether permeability measurements across different systems are governed by similar intermolecular interactions
- Results guide selection of appropriate high-throughput permeability screening methods for bRo5 compounds
- The approach speeds up drug candidate prioritization by ensuring consistent permeability assessment [1]
Protocol 3: IMHB Characterization in Solution
Objective: Characterize intramolecular hydrogen bonding in bRo5 compounds using spectroscopic and computational approaches.
Background: IMHB significantly influences the properties and behavior of bRo5 compounds. This protocol integrates experimental and computational methods to comprehensively characterize IMHB, providing insights into molecular chameleonicity and its impact on drug-like properties [43].
Materials:
- Spectroscopic Instruments: NMR spectrometer (preferably ≥ 400 MHz), FT-IR spectrometer
- Computational Resources: Workstation with quantum chemistry software (Gaussian, ORCA, or equivalent)
- Solvents: Deuterated solvents for NMR (CDCl₃, DMSO-d₆, etc.), spectroscopic-grade solvents for IR
- Reference Compounds: Compounds with known IMHB characteristics for method validation
Procedure:
- NMR Spectroscopy:
- Acquire ¹H NMR spectra in multiple deuterated solvents
- Perform variable temperature studies (typically 25-60°C)
- Measure two-bond deuterium isotope effects (TBDIE) on ¹³C chemical shifts
- Correct NH chemical shifts for ring current effects when aromatic substituents are present
Infrared Spectroscopy:
- Record IR spectra in appropriate solvents (non-polar, polar aprotic, polar protic)
- Identify OH or NH stretching frequencies and shifts
- Compare band positions in different environments
Computational Analysis:
- Perform conformational searching to identify low-energy conformers
- Conduct geometry optimization at appropriate theory level (e.g., DFT/B3LYP/6-311+G(d,p))
- Calculate hydrogen bond energies using Molecular Tailoring Approach or similar methods
- Analyze electron density topology using QTAIM or NCI approaches
Data Integration:
- Correlate experimental spectroscopic parameters with computed hydrogen bond strengths
- Assess solvent dependence of IMHB and conformational populations
- Evaluate potential for molecular chameleonicity
Data Interpretation:
- TBDIE on ¹³C chemical shifts provides sensitive measure of IMHB strength
- Red-shifts in OH/NH stretching frequencies indicate hydrogen bond formation
- Computational methods provide quantitative estimates of individual IMHB energies in complex systems
- Solvent-dependent changes indicate environment-responsive behavior (chameleonicity) [43]
The following workflow diagram illustrates the integrated approach to bRo5 compound property assessment:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for bRo5 Compound Research
| Category | Item | Specifications | Function/Application |
|---|---|---|---|
| Chromatographic Systems | Supelcosil LC-ABZ column | C18-based with embedded polar groups | Lipophilicity assessment of polar bRo5 compounds [1] [2] |
| Computational Software | MATLAB with BR analysis implementation | Custom scripts for interaction deconvolution | Interpreting balance of intermolecular forces in partitioning [1] |
| Spectroscopic Tools | NMR spectrometer | ≥400 MHz with variable temperature capability | IMHB characterization through TBDIE measurements [43] |
| Permeability Assays | PAMPA plates | Artificial membrane systems for high-throughput screening | BR-validated permeability assessment [1] |
| Computational Chemistry | Quantum chemistry packages | Gaussian, ORCA, or equivalent with DFT capabilities | Hydrogen bond energy calculations, conformational analysis [43] |
| Cell-Based Systems | Caco-2/MDCK cells | Validated for permeability studies | Reference standard for BR analysis of permeability methods [1] |
The strategic integration of bRo5 compound design with intramolecular hydrogen bonding optimization and Block Relevance analysis represents a powerful approach for addressing challenging drug targets. The protocols and strategies outlined in this document provide researchers with practical methodologies for navigating the complex property landscape of bRo5 compounds, enabling informed decision-making in drug discovery programs. By understanding and exploiting the unique characteristics of the bRo5 chemical space—including molecular chameleonicity, IMHB-mediated property modulation, and complex target engagement—medicinal chemists can expand the frontiers of druggability while maintaining acceptable pharmaceutical properties.
Optimizing HILIC Systems for Polar and Zwitterionic Molecules
Hydrophilic Interaction Liquid Chromatography (HILIC) has emerged as a powerful technique for analyzing polar and zwitterionic compounds that demonstrate poor retention in reversed-phase liquid chromatography (RPLC) [44]. This technique utilizes a polar stationary phase combined with a mobile phase containing a high percentage of organic solvent (typically >70% acetonitrile), where water acts as the stronger eluting solvent [44] [45]. The retention mechanism in HILIC is complex, involving a combination of partitioning, adsorption, and electrostatic interactions [46] [44]. For zwitterionic stationary phases, this complexity is further enhanced by their capacity to provide mixed-mode cation-exchange and anion-exchange interactions simultaneously [47].
The optimization of HILIC systems requires careful consideration of multiple chromatographic parameters, including stationary phase chemistry, mobile phase composition, buffer type and concentration, and pH. This application note provides detailed protocols for method development and optimization in HILIC, with particular emphasis on analyzing polar and zwitterionic molecules in pharmaceutical and environmental contexts. Furthermore, we frame this technical discussion within the broader research context of Block Relevance (BR) analysis, a computational tool that aids in deconvoluting the balance of intermolecular interactions governing chromatographic retention and lipophilicity assessment [1] [2].
Theoretical Background
HILIC Retention Mechanisms
The retention mechanism in HILIC is multifaceted, differing significantly from the relatively straightforward partitioning mechanism of RPLC. Three primary mechanisms contribute to retention in HILIC systems:
- Partitioning: Analyte partitioning between the bulk organic-rich mobile phase and a water-rich layer immobilized on the polar stationary phase surface [46] [44].
- Adsorption: Direct polar-polar interactions (e.g., hydrogen bonding, dipole-dipole) between analytes and the stationary phase ligands [46].
- Electrostatic Interactions: Ion-exchange interactions between charged analytes and ionizable groups on the stationary phase surface [47] [44].
The relative contribution of each mechanism depends on the specific stationary phase, analyte properties, and mobile phase conditions. For zwitterionic stationary phases, which contain both positively and negatively charged functional groups, electrostatic interactions play a particularly significant role [47].
Block Relevance Analysis in Chromatographic Method Development
Block Relevance (BR) analysis is a computational tool implemented in MATLAB that deconvolutes the balance of intermolecular interactions governing a given drug discovery-related phenomenon described by a QSPR/PLS model [1]. In the context of HILIC method development, BR analysis helps researchers:
- Identify the best chromatographic systems to provide reliable log P octanol/water (log Poct) surrogates.
- Interpret the lipophilicity and permeability of drug candidates by analyzing the contribution of different molecular interaction forces to chromatographic retention [1] [2].
- Determine the propensity of compounds to form intramolecular hydrogen bonds (IMHB) through analysis of Δlog Poct–tol values [3] [23].
By applying BR analysis, medicinal chemists can better interpret chromatographic data and make more informed decisions during method development and compound prioritization.
Critical Parameters for HILIC Optimization
Stationary Phase Selection
The selection of an appropriate stationary phase is crucial for successful HILIC separations. Different stationary phases offer distinct selectivity profiles and interaction mechanisms. The table below summarizes common HILIC stationary phases and their characteristics:
Table 1: Characteristics of Common HILIC Stationary Phases
| Stationary Phase Type | Chemical Functionality | Retention Mechanisms | Applications Notes |
|---|---|---|---|
| Bare Silica | Silanol groups (Si-OH) | Partitioning, hydrogen bonding, ion-exchange | Acidic silanols can cause peak tailing; sensitive to pH [44] |
| Diol | Neutral diol groups | Partitioning, hydrogen bonding | Good for neutral polar compounds; stable over wide pH [44] |
| Amide | Carbamoyl groups | Strong hydrogen bonding, partitioning | Excellent for sugars, peptides; very hydrophilic [44] |
| Zwitterionic Sulfobetaine | Sulfoalkylbetaine groups (+N and -SO3-) | Partitioning, ion-exchange, dipole-dipole | Simultaneous anion/cation exchange; self-shielding charges [47] [46] |
| Zwitterionic Phosphorylcholine | Phosphorylcholine groups | Partitioning, ion-exchange | Reversed charge order; thick water layer; peptide applications [46] |
Zwitterionic stationary phases are particularly valuable for separating polar and zwitterionic compounds due to their multimodal retention capabilities. Studies have demonstrated significant differences in the contribution of electrostatic interactions among different zwitterionic columns, with a general predominance of cation-exchange character over anion-exchange character observed across most tested columns [47]. The phosphorylcholine-type zwitterionic phase, which incorporates phosphoric acid groups instead of traditional sulfonic acid groups and reverses the order of charges in the ligand, has shown particular promise for retaining highly polar compounds [46].
Mobile Phase Composition
Mobile phase optimization is critical for achieving adequate retention and selectivity in HILIC. Key parameters to consider include:
- Organic Modifier Content: Typically 60-95% acetonitrile. Higher organic content increases retention of polar analytes. A minimum of 3% water is generally required to maintain the water-rich layer on the stationary phase [45].
- Buffer Type and Concentration: Volatile buffers like ammonium acetate or ammonium formate (5-20 mM) are recommended for MS compatibility. Buffer concentration significantly impacts peak shape and retention, particularly for ionic analytes [46] [45].
- pH Control: Mobile phase pH affects the ionization state of both analytes and stationary phase functional groups. Although pH is typically measured in the aqueous buffer before organic addition, the apparent pH in the final mobile phase will be different [45].
The following diagram illustrates the multifaceted retention mechanism in HILIC, which must be considered during mobile phase optimization:
Figure 1: HILIC Retention Mechanisms and Key Influencing Factors. Retention results from partitioning, adsorption, and electrostatic interactions, each influenced by different mobile phase parameters.
Method Optimization Using Design of Experiments (DoE)
A systematic approach to HILIC method optimization is crucial due to the complex interactions between chromatographic parameters. Experimental design (DoE) provides a structured methodology for evaluating multiple factors simultaneously while understanding their interaction effects [46].
Key factors to investigate in a HILIC DoE study include:
- Initial percentage of organic mobile phase
- Buffer concentration (e.g., 5-20 mM)
- Buffer type (e.g., ammonium acetate vs. ammonium formate)
- Mobile phase pH (within column stability limits)
- Column temperature
- Flow rate
- Gradient time
Studies have demonstrated that buffer type, flow rate, and initial percentage of organic mobile phase are often the most influential factors affecting retention, though the specific effects are closely related to the chemical and physicochemical properties of the analytes [46].
Experimental Protocols
Protocol 1: HILIC Method Development for Polar Molecules
Objective: Develop a robust HILIC method for the separation of polar emerging contaminants in reclaimed water.
Materials and Reagents:
- HILIC Column: Zwitterionic phosphorylcholine (SeQuant ZIC-cHILIC), 150 × 2.1 mm, 3.5 μm [46]
- Mobile Phase A: 5-20 mM ammonium acetate or ammonium formate in water, pH-adjusted with acetic or formic acid
- Mobile Phase B: Acetonitrile (LC-MS grade)
- Reference Standards: Target polar compounds (log DpH=7 range: -5.27 to 0.24)
- Instrumentation: LC-MS system with ESI source
Procedure:
- Column Equilibration: Equilibrate the column with initial mobile phase conditions (90-95% B) for at least 10-15 column volumes until stable baseline is achieved.
- Initial Scouting: Perform linear gradient from 95% to 60% B over 15-20 minutes at 0.3-0.5 mL/min flow rate and 25-40°C column temperature.
- DoE Optimization:
- Implement a full factorial or response surface design to evaluate critical factors (organic %, buffer concentration, pH).
- Assess responses (peak area, retention time, resolution) for all analytes.
- Use multivariate data analysis to identify optimal conditions.
- Final Method Conditions (example from literature [46]):
- Gradient: 95% to 60% B over 12 minutes
- Flow rate: 0.4 mL/min
- Temperature: 30°C
- Buffer: 10 mM ammonium acetate in water, pH 5.0
- Method Validation: Evaluate method performance for linearity, precision, accuracy, LOD, and LOQ.
Protocol 2: Zwitterionic Stationary Phase Characterization
Objective: Characterize the interaction properties and separation potential of zwitterionic stationary phases.
Materials and Reagents:
- Test Columns: Multiple zwitterionic columns (commercial and experimental)
- Reference Standards: Tanaka test mixture and proprietary compounds [47]
- Mobile Phase: Acetonitrile/water mixtures with appropriate buffers
Procedure:
- System Characterization:
- Apply the Tanaka testing protocol to evaluate column retentivity, hydrophobicity, hydrophilicity, and cation/anion-exchange characteristics [47].
- Analyze the test mixture at different pH values (e.g., 3.0, 4.7, 7.0) to assess pH-dependent behavior.
- Electrostatic Interaction Assessment:
- Measure retention of charged analytes at various buffer salt concentrations (e.g., 5-50 mM).
- Calculate the relative contribution of cation-exchange vs. anion-exchange mechanisms.
- Application Testing:
- Analyze tryptophan and its derivatives to evaluate selectivity for zwitterionic analytes.
- Test with cytochrome C tryptic digests to assess performance for complex samples.
- Data Analysis:
- Compare retention factors and selectivity factors between different columns.
- Determine the relative contribution of different retention mechanisms for each stationary phase.
Protocol 3: HILIC Method for Non-Target Screening
Objective: Develop a robust ZIC-HILIC-HRMS method for non-target screening of highly polar substances in industrial wastewater [48].
Materials and Reagents:
- HILIC Column: Zwitterionic (ZIC-HILIC), 150 × 2.0 mm, 3.5 μm
- Mobile Phase A: 10 mM ammonium acetate in water, pH ~6.8 (adjusted with acetic acid)
- Mobile Phase B: Acetonitrile (LC-MS grade)
- Quality Control Samples: Mix of 20 reference substances with log D values from -7 to 2 at pH 7.4
Procedure:
- Sample Preparation: Dilute wastewater samples with ACN (typically 1:10 for influent, undiluted for effluent) [48].
- Chromatographic Conditions:
- Gradient: 90% B to 50% B over 15 minutes
- Flow rate: 0.3 mL/min
- Column temperature: 30°C
- Injection volume: 5 μL
- MS Parameters:
- Ionization: ESI positive/negative switching
- Mass range: 50-1000 m/z
- Resolution: >70,000
- Method Validation:
- Assess repeatability and reproducibility (target %RSD of intensity <6%, retention time <1%).
- Evaluate linearity (R² > 0.99 for 75% of substances).
- Determine LOD (typically 0.1-40 μg/L for tested compounds).
- Data Processing:
- Perform principal component analysis (PCA) to identify patterns in wastewater content.
- Monitor removal efficiency in wastewater treatment processes.
The following workflow summarizes the comprehensive approach to HILIC method development and application:
Figure 2: Comprehensive HILIC Method Development Workflow. The systematic approach includes stationary phase screening, initial scouting, DoE optimization, and validation before application to various sample types.
Research Toolkit
Table 2: Essential Research Reagent Solutions for HILIC Method Development
| Item | Specifications | Function/Application |
|---|---|---|
| Zwitterionic HILIC Columns | ZIC-cHILIC (phosphorylcholine), ZIC-HILIC (sulfobetaine), 150 × 2.1 mm, 3.5 μm | Primary separation media for polar/zwitterionic compounds [46] [48] |
| Mobile Phase Buffers | Ammonium acetate, ammonium formate (LC-MS grade), 5-20 mM in water | Volatile buffers for MS compatibility; control ionic interactions [46] [45] |
| Organic Modifiers | Acetonitrile (LC-MS grade) | Primary organic component in HILIC mobile phases [46] [44] |
| pH Adjustors | Formic acid, acetic acid, ammonium hydroxide (LC-MS grade) | Fine-tuning pH for ionization control of analytes/stationary phase [46] [45] |
| Reference Standards | Tanaka test mixture, polar compounds with known log D values | System characterization and method validation [47] [48] |
Applications and Data Analysis
Application to Polar Emerging Contaminants
HILIC methods employing zwitterionic stationary phases have been successfully applied to the analysis of Persistent and Mobile Organic Contaminants (PMOCs) in reclaimed water [46]. These methods enable the simultaneous determination of diverse polar compounds including pesticides, artificial sweeteners, pharmaceuticals, and central nervous system stimulants with log DpH=7 values ranging from -5.27 to 0.24.
Optimized methods have demonstrated acceptable figures of merit, with recoveries ranging from 49% to 100% for most analytes and satisfactory precision (RSD <10% for all analytes) [46]. The successful application to real-world water samples highlights the practical utility of well-optimized HILIC methods for environmental monitoring.
Data Interpretation Using Block Relevance Analysis
Block Relevance (BR) analysis provides a powerful framework for interpreting chromatographic data in terms of underlying molecular interactions. When applied to HILIC retention data, BR analysis can:
- Identify the dominant intermolecular interactions governing retention for different classes of compounds.
- Facilitate the conversion of chromatographic indices to log P values by ensuring the chromatographic system expresses the same balance of intermolecular solute/system forces as the reference partition system [1] [2].
- Support the use of Δlog Poct–tol as a molecular descriptor for hydrogen bond donor properties of solutes, which is valuable for interpreting intramolecular hydrogen bonding [3].
By applying BR analysis, researchers can move beyond empirical method development to a more mechanistic understanding of retention behavior in HILIC systems.
The optimization of HILIC systems for polar and zwitterionic molecules requires careful consideration of multiple chromatographic parameters, with zwitterionic stationary phases offering particularly valuable multimodal retention mechanisms for challenging separations. The implementation of systematic optimization approaches, including Design of Experiments, enables efficient method development for complex applications in pharmaceutical analysis and environmental monitoring.
When framed within the context of Block Relevance analysis, HILIC method development transitions from an empirical process to a more rational approach based on understanding the fundamental intermolecular interactions governing retention. This integrated perspective enhances our ability to develop robust analytical methods for polar and zwitterionic compounds while simultaneously generating valuable physicochemical data to support drug discovery and environmental chemistry research.
Addressing Discrepancies Between Different Lipophilicity Measurement Methods
Lipophilicity, most commonly quantified as the logarithm of the partition coefficient (log P) for the neutral form of a compound or the distribution coefficient (log D) at a specific pH, is a fundamental physicochemical property in drug design and development. It profoundly influences a compound's absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile [49] [50]. The accurate assessment of lipophilicity is therefore critical for selecting viable drug candidates. However, researchers often encounter significant discrepancies when comparing lipophilicity values obtained through different measurement techniques. These discrepancies arise from the varied physicochemical principles and experimental conditions underpinning each method. This Application Note examines the sources of these discrepancies and provides detailed protocols and analytical frameworks to aid in their interpretation, contextualized within Block Relevance analysis for robust lipophilicity assessment.
Discrepancies between lipophilicity measurements stem from fundamental differences in methodological approaches: computational algorithms versus experimental techniques, and variations among experimental platforms themselves.
Computational (in silico) Methods
In silico methods predict log P based on the molecular structure using diverse algorithms. Different software packages employ distinct calculation approaches, such as atom-based, fragment-based, topological, or physics-based methods, leading to varying predictions for the same molecule [51]. For instance, a study on neuroleptics and their derivatives reported values from multiple algorithms, including ALOGPS, XLOGP3, and iLOGP, which often showed variations for a single compound [49]. The consensus log P, calculated as the arithmetic mean of several diverse predictors, is often used to improve reliability [51].
Experimental Methods
Experimental methods measure lipophilicity through a compound's behavior in partitioned systems, but are influenced by the specific conditions of the assay.
- Shake-Flask Method: This is the classical benchmark method for measuring partition coefficients [52]. It involves directly measuring the distribution of a compound between immiscible aqueous and organic (typically n-octanol) phases. It can be slow, compound-intensive, and is susceptible to impurities and degradation products [53].
- Chromatographic Methods: These techniques infer lipophilicity from a compound's retention time or retention factor on a non-polar stationary phase.
- Reverse-Phase High-Performance Liquid Chromatography (RP-HPLC): This method is recommended by IUPAC as equivalent to the shake-flask method [52]. The chromatographic hydrophobicity index (CHI) derived from RP-HPLC shows good correlation with measured log D values [53]. The choice of stationary phase (e.g., C8, C18, Immobilized Artificial Membrane - IAM) and mobile phase organic modifier (e.g., methanol, acetonitrile) significantly impacts the retention behavior and thus the derived lipophilicity parameter [53] [52].
- Reverse-Phase Thin-Layer Chromatography (RP-TLC/HPTLC): A simpler and faster chromatographic technique where lipophilicity is expressed as the RMW parameter [49] [52]. The stationary phase (e.g., RP-2, RP-8, RP-18) and the type of organic modifier (e.g., acetone, acetonitrile, methanol, 1,4-dioxane) are key variables [49].
The underlying principle of "Block Relevance" is evident here: each method (computational block or experimental block) provides a different, albeit relevant, perspective on the lipophilic character of a molecule. A computational method might describe an "ideal" partitioning system, while a chromatographic method reflects lipophilicity in the context of specific chemical interactions with the stationary phase.
Quantitative Comparison of Lipophilicity Parameters
The following tables summarize lipophilicity data from recent studies, illustrating the variations observed across different methodologies.
Table 1: Comparison of Calculated log P Values for Neuroleptics from Different Algorithms [49]
| Compound | ALOGPS | iLOGP | XLOGP3 | WLOGP | MLOGP | Consensus log P |
|---|---|---|---|---|---|---|
| Fluphenazine | 4.77 | 4.63 | 5.00 | 4. | 1.81 | 4.10 |
| Triflupromazine | 5.38 | 5.16 | 5.52 | 5.22 | 3.46 | 4.95 |
| Trifluoperazine | 4.87 | 5.08 | 5.53 | 5.19 | 3.28 | 4.79 |
| Flupentixol | 4.27 | 4.30 | 4.73 | 4.45 | 2.99 | 4.15 |
| Zuclopenthixol | 4.80 | 4.85 | 5.48 | 5.01 | 3.53 | 4.73 |
Table 2: Experimental Chromatographic Lipophilicity Parameters (RMW) for Neuroleptics on Different RP-TLC Stationary Phases [49]
| Compound | RP-2/F254 (Acetone) | RP-2/F254 (Acetonitrile) | RP-2/F254 (1,4-Dioxane) | RP-8/F254 (Acetone) | RP-8/F254 (Acetonitrile) | RP-8/F254 (1,4-Dioxane) | RP-18/F254 (Acetone) | RP-18/F254 (Acetonitrile) | RP-18/F254 (1,4-Dioxane) |
|---|---|---|---|---|---|---|---|---|---|
| Fluphenazine | 1.68 | 1.21 | 1.83 | 2.13 | 1.58 | 2.12 | 2.52 | 1.81 | 2.40 |
| Triflupromazine | 2.36 | 1.91 | 2.42 | 2.67 | 2.22 | 2.64 | 3.16 | 2.58 | 3.02 |
| Trifluoperazine | 1.88 | 1.43 | 1.96 | 2.31 | 1.79 | 2.25 | 2.74 | 2.03 | 2.61 |
| Flupentixol | 1.61 | 1.15 | 1.73 | 2.06 | 1.52 | 2.02 | 2.48 | 1.78 | 2.34 |
| Zuclopenthixol | 2.08 | 1.61 | 2.10 | 2.48 | 1.93 | 2.39 | 2.92 | 2.19 | 2.78 |
Table 3: Lipophilicity Parameters (log kw) for 1,3,4-Thiadiazoles Determined by HPLC with Different Stationary Phases [52]
| Compound | C8/MeOH | C8/ACN | C18/ACN | IAM/pH 7.4 | Chol/pH 7.4 |
|---|---|---|---|---|---|
| 4 | 1.71 | 1.46 | 1. | 0.90 | 1.10 |
| 6 | 1.54 | 1.32 | 1.48 | 1.16 | 1.38 |
| 11 | 2.37 | 2.09 | 2.38 | 1.67 | 2.08 |
| 13 | 1.26 | 1.04 | 1.13 | 0.92 | 1.11 |
| 15 | 3.02 | 2.79 | 3.16 | 2.21 | 2.76 |
Detailed Experimental Protocols
Protocol: Determining Lipophilicity by RP-TLC
This protocol outlines the procedure for determining the RMW parameter as a measure of lipophilicity [49].
4.1.1 Materials and Equipment
- TLC Plates: Commercially available RP-2 F254, RP-8 F254, and RP-18 F254 plates.
- Organic Modifiers: HPLC-grade acetone, acetonitrile, and 1,4-dioxane.
- Sample Solution: Dissolve test compounds in a volatile solvent (e.g., methanol) at a concentration of ~1 mg/mL.
- Development Chamber: A standard, flat-bottomed TLC chamber with a lid.
- Micropipettes: For sample application.
- UV Lamp: A lamp with a wavelength of 254 nm for visualizing spots.
4.1.2 Procedure
- Mobile Phase Preparation: Prepare mobile phases with varying volume fractions (φ) of the organic modifier in water (e.g., 50%, 60%, 70%, 80%). Ensure thorough mixing.
- Sample Application: Using a micropipette, spot 1-2 µL of each sample solution onto the baseline of the TLC plate (~1 cm from the bottom). Allow the solvent to evaporate completely.
- Chromatogram Development: Pour the mobile phase into the chamber to a depth of ~0.5 cm and allow saturation for 15-20 minutes. Place the spotted TLC plate in the chamber and develop until the solvent front has migrated 6-8 cm from the origin.
- Drying and Visualization: Remove the plate from the chamber, mark the solvent front, and allow it to air-dry in a fume hood. Visualize the spots under UV light at 254 nm and circle them with a pencil.
- Data Calculation:
- Measure the distance from the start line to the center of the spot (DS) and from the start line to the solvent front (DF).
- Calculate the retention factor (RF) for each spot: RF = DS / DF.
- Calculate the RM value for each spot: RM = log(1/R_F - 1).
- Determining RMW: For each compound and chromatographic system, plot the RM values against the volume fraction (φ) of the organic modifier in the mobile phase. Perform linear regression. The extrapolated RM value at φ = 0 (the y-intercept) is the RMW lipophilicity parameter: RM = RMW + bφ.
Protocol: Determining Lipophilicity by RP-HPLC
This protocol describes the measurement of the log kw parameter using an isocratic RP-HPLC method [53] [52].
4.2.1 Materials and Equipment
- HPLC System: Equipped with a pump, autosampler, column thermostat, and detector (e.g., UV/VIS or DAD).
- HPLC Columns: C8, C18, IAM, or cholesterol-bonded stationary phases.
- Mobile Phase: Aqueous buffer (e.g., phosphate buffer pH 7.4) and organic modifiers (methanol, acetonitrile). All solvents should be HPLC grade.
- Test Compounds: Reference standards and samples of interest.
4.2.2 Procedure
- Mobile Phase Preparation: Prepare a series of isocratic mobile phases with varying volume fractions (φ) of organic modifier in the aqueous buffer (e.g., 60%, 70%, 80%, 90% organic). Degas all mobile phases before use.
- HPLC System Setup: Equilibrate the chosen column with the mobile phase at a constant temperature (e.g., 25°C) and a stable flow rate (e.g., 1.0 mL/min) until a stable baseline is achieved.
- Injection and Analysis: Inject a small volume (e.g., 10 µL) of each sample solution. Record the retention time (tR) for each compound. Also, record the void time (t0) using a non-retained compound (e.g., uracil or potassium iodide).
- Data Calculation:
- Calculate the retention factor (k) for each compound: k = (tR - t0) / t_0.
- Calculate the logarithm of the retention factor: log k.
- Determining log kw: For each compound and chromatographic system, plot the log k values against the volume fraction (φ) of the organic modifier. Perform linear regression. The extrapolated log k value at φ = 0 is the log kw lipophilicity parameter: log k = log kw + bφ.
Visualizing Method Selection and Data Integration
The following diagrams illustrate the workflow for method selection based on Block Relevance and the process for integrating and interpreting multi-method data.
Lipophilicity Method Selection Logic
Multi-Method Data Integration Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials and Tools for Lipophilicity Assessment
| Item | Function/Description | Example Use |
|---|---|---|
| RP-TLC Plates (C8, C18) | Silica gel plates bonded with octyl or octadecyl chains; used for rapid lipophilicity screening. | Determining RMW values with different organic modifiers [49] [52]. |
| RP-HPLC Columns (C8, C18, IAM) | Columns with hydrocarbon-coated or biomimetic (Immobilized Artificial Membrane) stationary phases. | Measuring log kw under isocratic conditions; IAM phases model cell membrane interactions [53] [52]. |
| ACD/LogP & ChemSketch | Freeware for predicting log P based on chemical structure and for drawing molecular structures. | Rapid in silico estimation of lipophilicity during compound design [54]. |
| SwissADME Web Tool | A free online tool that provides multiple log P predictions and a consensus value. | Accessing diverse algorithms (XLOGP3, iLOGP, etc.) for a consensus view [51]. |
| Methanol & Acetonitrile | Common organic modifiers used in RP-TLC and RP-HPLC mobile phases. | Creating binary eluent systems with water/buffer to modulate compound retention [49] [52]. |
| 1,4-Dioxane & Acetone | Alternative organic modifiers for chromatographic systems. | Useful for extending the range of measurable lipophilicity, especially in RP-TLC [49]. |
Discrepancies between lipophilicity measurement methods are not merely experimental noise but contain valuable information about the multifaceted nature of molecular lipophilicity. A Block Relevance analysis framework, which treats data from different methodological blocks (e.g., computational, C18-HPLC, IAM-HPLC) as distinct yet complementary, allows for a more nuanced and biologically relevant interpretation. By employing a combination of the protocols and analytical tools outlined in this note, researchers can transform methodological discrepancies into a deeper understanding of their compounds, ultimately guiding the more effective design of drug candidates with optimal ADMET properties.
Guidelines for Safer Method Selection in Permeability Studies
Permeability studies are fundamental in drug development, providing critical data on a compound's ability to cross biological barriers and reach its intended site of action. The reliability of these studies hinges on the careful selection and validation of experimental methods. Method suitability provides a generalized approach to standardize and validate permeability models within laboratories, accounting for intra- and inter-laboratory variability while accommodating technological improvements [55]. This document establishes guidelines for safer method selection within the context of Block Relevance analysis for lipophilicity assessment, enabling researchers to generate dependable permeability data for informed decision-making during drug discovery and development.
Theoretical Foundation: Permeability and Lipophilicity
The permeability of a drug molecule across biological membranes is intrinsically linked to its lipophilicity, commonly quantified as the logarithm of the n-octanol/water partition coefficient (LogP). According to Fick's first law, passive drug flux (Jr) is proportional to membrane permeability (Pm) and drug concentration at the membrane surface (Ci) [56]. Passive diffusion, the primary absorption mechanism for most commercial drugs (80-95%), favors compounds with balanced lipophilicity, lower polarity, and smaller molecular weight [56] [57].
The Biopharmaceutics Classification System (BCS) categorizes drugs based on solubility and permeability, providing a framework for predicting absorption [56]. Method selection must therefore enable accurate permeability classification to distinguish between high permeability (HP, fa ≥ 90%) and low permeability (LP, fa < 90%) drug substances [55].
Table 1: Biopharmaceutics Classification System (BCS) Framework
| Class | Solubility | Permeability | Example Drugs |
|---|---|---|---|
| I | High | High | Acyclovir, captopril, abacavir |
| II | Low | High | Atorvastatin, diclofenac, ciprofloxacin |
| III | High | Low | Cimetidine, atenolol, amoxicillin |
| IV | Low | Low | Furosemide, chlorthalidone, methotrexate |
Adapted from [56]
Permeability Models: Comparative Analysis
Model Categories and Characteristics
Multiple experimental models exist for assessing drug permeability, each with distinct advantages, limitations, and appropriate applications. Selection depends on research stage, throughput requirements, and physiological relevance.
Table 2: Permeability Assay Comparison for Method Selection
| Assay Type | Key Features | Advantages | Limitations | Suitability for Lipophilicity Assessment |
|---|---|---|---|---|
| In Silico | Computational prediction using LogP, molecular descriptors, ML, MD simulations [56] [58] | High throughput, cost-effective early screening, handles virtual compounds | Limited to passive diffusion, accuracy depends on model training | High - Directly calculates lipophilicity parameters; core for Block Relevance analysis |
| Artificial Membranes (PAMPA) | Biomimetic membranes impregnated with lipids [55] [57] | High throughput, reproducible, models passive transcellular diffusion [55] [59] | Lacks transporters & metabolic enzymes; limited physiological relevance [55] | High - Excellent for assessing pure physicochemical permeability driven by lipophilicity |
| Cell-Based Monolayers (Caco-2, MDCK) | Human/animal cell lines forming differentiated monolayers [55] [59] | Models transcellular/paracellular diffusion, active transport, and efflux; human-relevant pathways [55] | Extended cultivation (Caco-2), inter-lab variability, low transporter expression [55] [59] | Medium-High - Reveals interplay between lipophilicity and biological transport mechanisms |
| Ex Vivo Tissue Models (Everted sac, Using chamber) | Actual intestinal/ mucosal tissues in controlled systems [55] [60] | Retains native tissue architecture, metabolic activity, and regional differences [55] | Limited tissue viability, suboptimal stirring conditions, biological variability [55] | Medium - Provides intact biological environment to contextualize lipophilicity |
| In Situ Perfusion | Perfusion of intestinal segments in living animals [55] | Retains blood flow, innervation, and in vivo physiology [55] | Low throughput, requires animal surgery, ethically demanding [55] | Medium-Low - Confirms in vivo relevance but lower throughput for screening |
Strategic Model Selection Framework
Safer method selection requires aligning model capabilities with specific research questions. The following workflow provides a systematic approach for selecting appropriate permeability assays based on study objectives and compound characteristics:
Experimental Protocols for Method Suitability
Method Suitability Framework
Implementing method suitability ensures reliability and reproducibility of permeability data. This framework involves three critical stages [55]:
Stage 1: Method Development and Standardization
- Assay Protocol Establishment: Define and optimize all experimental parameters including buffer composition, pH, temperature, incubation time, and sampling intervals [55]
- Acceptance Criteria Definition: Establish specifications for barrier integrity (e.g., transepithelial electrical resistance), viability markers, and reference compound permeability [55]
- Functional Characterization: Assess presence of functional active transporters and efflux mechanisms using known substrates [55]
Stage 2: Demonstrating Assay Suitability
- IVIVC Establishment: Create rank-order relationship between experimental permeability values (Papp or Peff) and human intestinal absorption extent using model drugs spanning absorption ranges (<50%, 50-89%, ≥90%) [55]
- HP-IS Selection: Identify a highly permeable internal standard with permeability near the high/low class boundary to facilitate test drug classification [55]
Stage 3: Permeability Classification
- Reference Standards: Implement LP and HP drugs, transporter substrates, and integrity markers (e.g., mannitol, Lucifer yellow) in each study [55]
- Quality Control: Regularly monitor standard compound performance to ensure assay reproducibility and reliability [55]
Detailed Protocol: Caco-2 Permeability Assay
Principle: Human colorectal adenocarcinoma cells (Caco-2) differentiate into polarized monolayers resembling intestinal epithelium, expressing transporters, tight junctions, and efflux mechanisms [59].
Materials:
- Caco-2 cell line (ATCC)
- DMEM with 10% FBS, 1% non-essential amino acids, and 1% penicillin-streptomycin
- Transwell inserts (0.4 μm pore size, 12-well format)
- Transport buffer (HBSS with 25 mM glucose and 10 mM HEPES, pH 7.4)
- Test compounds and reference standards (e.g., propranolol HP, atenolol LP)
- Integrity markers (e.g., Lucifer yellow, mannitol)
- HPLC system with UV/fluorescence detection or LC-MS/MS
Procedure:
- Cell Culture and Seeding:
- Maintain Caco-2 cells in complete medium at 37°C, 5% CO₂
- Seed cells on Transwell inserts at density of 1×10⁵ cells/cm²
- Culture for 21-28 days with medium changes every 2-3 days
Monolayer Integrity Validation:
- Measure transepithelial electrical resistance (TEER) ≥300 Ω·cm²
- Confirm paracellular marker flux (e.g., Lucifer yellow Papp < 2×10⁻⁶ cm/s)
Transport Experiment:
- Pre-incubate monolayers with transport buffer for 20 min at 37°C
- Add test compound (typically 10-100 μM) to donor compartment (apical for A-B, basolateral for B-A)
- Sample receiver compartment at intervals (e.g., 30, 60, 90, 120 min)
- Maintain sink conditions (<10% compound accumulation in receiver)
- Include reference standards in each experiment
Sample Analysis and Calculations:
- Analyze samples using HPLC or LC-MS/MS
- Calculate apparent permeability: Papp = (dQ/dt) / (A × C₀)
- dQ/dt = transport rate (mol/s)
- A = membrane surface area (cm²)
- C₀ = initial donor concentration (mol/mL)
Acceptance Criteria:
- Reference standard Papp values within historical laboratory range (e.g., propranolol Papp > 10×10⁻⁶ cm/s, atenolol Papp < 1×10⁻⁶ cm/s)
- Integrity marker recovery <1% of applied dose
- TEER values maintained within 20% of pre-experiment values
Detailed Protocol: PAMPA for Lipophilicity Assessment
Principle: Parallel Artificial Membrane Permeability Assay uses artificial membranes to model passive transcellular permeability, ideal for Block Relevance analysis of lipophilicity- permeability relationships [61].
Materials:
- Multi-well filter plates (e.g., 96-well format)
- Artificial membrane lipid solution (e.g., Lecithin in n-dodecane)
- Test compounds dissolved in DMSO (<1% final)
- Donor and acceptor buffer (e.g., PBS pH 7.4 or physiologically relevant pH)
- UV-compatible plates and plate reader
Procedure:
- Membrane Formation:
- Add lipid solution to filter plate and incubate to form artificial membrane
- Verify membrane uniformity visually
Permeability Experiment:
- Add compound solution to donor compartment
- Fill acceptor compartment with buffer
- Seal plates to prevent evaporation
- Incubate with gentle agitation for 2-16 hours at 25°C
Analysis and Calculations:
- Measure compound concentration in donor and acceptor compartments
- Calculate permeability: Papp = -ln(1 - Cₐ/Cₑq) / [A × (1/Vd + 1/Va) × t]
- Cₐ = acceptor concentration at time t
- Cₑq = equilibrium concentration
- A = membrane area
- Vd, Va = donor and acceptor volumes
- t = incubation time
The Scientist's Toolkit: Essential Research Reagents
Table 3: Research Reagent Solutions for Permeability Studies
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Caco-2 Cell Line | Human intestinal epithelial model | Forms polarized monolayers with tight junctions, transporters; 21-28 day differentiation [59] |
| MDCK Cell Line | Canine kidney epithelial model | Faster differentiation (7-10 days), lower transporter expression; suitable for transfection studies [59] [58] |
| HT29-MTX Cell Line | Human intestinal goblet cell model | Mucin-producing; used in co-culture with Caco-2 to incorporate mucus layer [59] |
| Transwell Inserts | Permeable supports for cell culture | Polycarbonate or polyester membranes (0.4-3.0 μm pores) in 6- to 96-well formats |
| PAMPA Membrane Lipids | Artificial membrane formation | Lecithin, cholesterol, n-octanol mixtures to mimic specific biological barriers [61] [57] |
| Reference Compounds | Assay standardization and validation | HP: Propranolol, metoprolol, antipyrine; LP: Atenolol, ranitidine; Transporter substrates [55] |
| Integrity Markers | Barrier function assessment | Paracellular flux markers: Lucifer yellow, mannitol, PEG 4000; non-permeability confirms tight junction integrity [55] |
| Franz Diffusion Cells | Static diffusion system | Vertical cells for artificial membranes or excised tissues; temperature-controlled water jacket [57] |
Data Interpretation and Risk Mitigation
Acceptance Criteria and Quality Control
Robust permeability classification requires strict quality control measures throughout experimentation. Implementation of systematic acceptance criteria minimizes variability and enhances data reliability [55].
Critical Quality Metrics:
- Barrier Integrity: TEER values ≥300 Ω·cm² for Caco-2; paracellular marker Papp < 2×10⁻⁶ cm/s
- Reference Standard Ranges: Laboratory-specific historical ranges for HP and LP compounds
- Assay Precision: Coefficient of variation <15-20% for replicate measurements
- Recovery Mass Balance: 100±15% of applied compound accounted for in donor, receiver, and membrane compartments
Data Normalization and Benchmarking
Permeability values demonstrate significant inter-laboratory variability due to methodological differences. Cross-system comparisons should utilize data normalized to well-characterized reference standards rather than absolute permeability values [55] [58].
Benchmarking Protocol:
- Internal Standard Normalization: Express test compound permeability relative to HP-IS
- Classification Criteria: Test drug Papp ≥ HP-IS Papp classifies as highly permeable [55]
- Inter-laboratory Comparison: Use common reference compounds to facilitate data comparison across research sites
Safer method selection in permeability studies requires systematic implementation of standardized protocols, appropriate model selection based on research objectives, and rigorous quality control measures. The integration of Block Relevance analysis for lipophilicity assessment enhances prediction accuracy by focusing on the fundamental physicochemical drivers of passive permeability. Through adherence to methodological suitability frameworks, employment of appropriate reference standards, and establishment of laboratory-specific acceptance criteria, researchers can generate reliable permeability data that effectively informs drug discovery and development decisions, ultimately contributing to more efficient development of effective therapeutic agents.
Validation and Comparative Analysis: How BR Analysis Stacks Up
Benchmarking Against Traditional and AI-Based Molecular Property Prediction Methods
Molecular property prediction is a cornerstone of modern drug discovery, enabling researchers to prioritize compounds with desirable pharmacokinetic and safety profiles. Lipophilicity, quantitatively expressed as the distribution coefficient at pH 7.4 (logD7.4), profoundly influences a drug's absorption, distribution, metabolism, excretion, and toxicity (ADMET). Accurate prediction of this property is therefore crucial for successful drug development [62].
This application note provides a structured framework for benchmarking traditional and Artificial Intelligence (AI)-based methods for molecular property prediction, with a specific focus on lipophilicity within the context of Block Relevance (BR) analysis. BR analysis is a computational tool that deconvolutes the balance of intermolecular interactions governing a drug discovery-related phenomenon described by a QSPR/PLS model, thereby aiding in the interpretation of lipophilicity and permeability data [1]. The protocols outlined herein are designed for researchers, scientists, and drug development professionals seeking to evaluate and implement these predictive methodologies.
Background and Significance
Molecular property prediction techniques generally fall into three categories based on their representation of the chemical structure: fixed representations, learned representations, and 3D descriptions.
Fixed representations, such as molecular fingerprints (e.g., Extended-Connectivity Fingerprints, ECFP) and pre-calculated molecular descriptors (e.g., RDKit2D), are hand-crafted features that signify the presence of specific structural patterns [63]. These are typically used with traditional machine learning models like Support Vector Regression (SVR) and LASSO [64].
In contrast, AI-based methods leverage learned representations through deep learning. These include Graph Neural Networks (GNNs) that operate directly on molecular graphs, and sequence models that process Simplified Molecular Input Line Entry System (SMILES) strings [63] [65]. These techniques can automatically learn relevant features from the data.
A third approach moves beyond commonly used 2D descriptors to a more physically relevant 3D molecular description, which can provide a more complete picture of molecular shape and electrostatics [66].
A critical challenge in the field, particularly for AI methods, is the limited availability of high-quality experimental data, which can restrict model generalizability [67] [62]. Furthermore, dataset size has been shown to be essential for representation learning models to excel, and their performance can be limited in low-data regimes [63]. Techniques like multi-task learning (MTL) and transfer learning are being actively developed to mitigate these data scarcity issues [67] [62].
Benchmarking Data and Quantitative Comparison
A rigorous benchmark requires diverse datasets and standardized evaluation metrics. The following tables summarize key datasets and a performance comparison of various methods.
Table 1: Key Molecular Property Datasets for Benchmarking
| Dataset Name | Primary Property | Size (Molecules) | Key Characteristics |
|---|---|---|---|
| LIPOPEP [64] | logD7.4 | 243 | Short, linear natural peptides |
| AZ (in-house) [64] | logD7.4 | 800 | Complex peptide derivatives and mimetics |
| DB29-data (ChEMBL) [62] | logD7.4 | Not Specified | Curated from ChEMBLdb29 (shake-flask, chromatographic, potentiometric data) |
| Lipophilicity (Lipo) [65] | Octanol/water distribution coeff. | 4,200 | Part of the MoleculeNet benchmark suite |
| ESOL [65] | Water Solubility | 1,128 | Estimated water solubility |
| FreeSolv [65] | Hydration Free Energy | 642 | Hydration free energy in water |
| QM9 [65] | Quantum Mechanical Properties | 130,831 | Includes dipole moment, spatial information |
Table 2: Performance Comparison of Representative Prediction Models
| Model / Technique | Representation | Dataset | Performance (RMSE) | Key Context |
|---|---|---|---|---|
| SVR(Lasso) [64] | Fixed Descriptors | LIPOPEP | 0.39 | Linear peptide model; 90.6% predictions within ±0.5 log units |
| LASSO [64] | Fixed Descriptors | LIPOPEP | 0.54 | Linear peptide model; 73.4% predictions within ±0.5 log units |
| RTlogD [62] | GNN + Transfer Learning | DB29-data | Superior to common algorithms | Integrates retention time, pKa, logP; outperformed tools like ADMETlab2.0 |
| ACS (GNN) [67] | Molecular Graph | ClinTox, SIDER, Tox21 | Matched or surpassed state-of-the-art | Effective in low-data regimes; mitigates negative transfer in multi-task learning |
| Quantized GNN [65] | Molecular Graph | ESOL, FreeSolv, Lipo, QM9 | Performance maintained (8-bit) | Reduces memory/computational cost; aggressive 2-bit quantization degrades performance |
Experimental Protocols
Protocol 1: Benchmarking Fixed Representation Models
This protocol details the steps for building and evaluating a lipophilicity model using fixed molecular representations and traditional machine learning.
4.1.1 Research Reagent Solutions
- Software Environment: Python with RDKit for descriptor/fingerprint calculation and Scikit-learn for machine learning models.
- Molecular Descriptors: Use RDKit to calculate a set of 2D descriptors (e.g., MolWt, MolLogP, NumHDonors, NumHAcceptors) [63].
- Molecular Fingerprints: Generate Extended-Connectivity Fingerprints (ECFP4 or ECFP6) with a bit vector size of 1024 or 2048 [63].
- Machine Learning Models:
- LASSO (Least Absolute Shrinkage and Selection Operator): A linear model that performs feature selection via L1 regularization. Tune the
αparameter to minimize cross-validation error [64]. - SVR (Support Vector Regression): A non-linear kernel-based method. Use a Gaussian kernel and optimize hyperparameters
Candγ[64].
- LASSO (Least Absolute Shrinkage and Selection Operator): A linear model that performs feature selection via L1 regularization. Tune the
4.1.2 Step-by-Step Procedure
- Data Preparation: Assemble a dataset of molecules with experimental logD7.4 values. Pre-process the data by removing duplicates and correcting errors [62].
- Descriptor Calculation: For each molecule in the dataset, compute molecular descriptors and/or fingerprints using RDKit.
- Feature Preprocessing: Normalize the calculated descriptors to a common scale (e.g., zero mean and unit variance).
- Model Training: Split the data into training (e.g., 80%) and test (e.g., 20%) sets. Train the LASSO and SVR models on the training set. Use cross-validation on the training set to select optimal hyperparameters.
- Model Evaluation: Predict logD7.4 values for the held-out test set and calculate performance metrics, including Root-Mean-Square Error (RMSE) and the percentage of predictions within ±0.5 log units of the experimental value.
Protocol 2: Benchmarking Graph Neural Network Models
This protocol outlines the process for training and evaluating a GNN for property prediction, incorporating advanced techniques to handle data scarcity.
4.2.1 Research Reagent Solutions
- Software Environment: PyTorch Geometric or Deep Graph Library for GNN implementation.
- GNN Architecture:
- Advanced Training Schemes:
- ACS (Adaptive Checkpointing with Specialization): A training scheme for multi-task GNNs that mitigates negative transfer by checkpointing the best model parameters for each task individually [67].
- Knowledge Transfer Techniques:
4.2.2 Step-by-Step Procedure
- Data Preparation and Splitting: Curate a logD7.4 dataset. For a more realistic assessment, use a time-split or scaffold-split to separate training and test data, ensuring that the test set contains structurally novel molecules [67] [62].
- Graph Representation: Convert each molecule's SMILES string into a graph representation where atoms are nodes and bonds are edges. Initialize node features with atom properties (e.g., atomic number, degree) and edge features with bond properties (e.g., bond type) [63].
- Model Configuration: Choose a GNN architecture (e.g., GIN, GCN). For MTL, implement an ACS scheme that uses a shared GNN backbone with task-specific prediction heads.
- Training and Validation:
- For Single-Task Learning: Train the GNN to minimize the prediction error (e.g., RMSE) on the training set, using a validation set for early stopping.
- For Multi-Task Learning with ACS: Train the shared backbone and task-specific heads simultaneously. Monitor the validation loss for each task independently and save a checkpoint (backbone + head) whenever a task achieves a new minimum validation loss [67].
- Evaluation: Report the performance of the final model(s) on the test set for each task.
Protocol 3: Applying Block Relevance (BR) Analysis
This protocol describes how to integrate BR analysis to interpret the lipophilicity prediction model and guide the selection of the most appropriate experimental measurement system.
4.3.1 Research Reagent Solutions
- BR Analysis Software: Implementation of BR analysis in MATLAB [1].
- Chromatographic Systems: A variety of HPLC columns (e.g., Supelcosil LC-ABZ) and conditions for measuring chromatographic indices [2].
4.3.2 Step-by-Step Procedure
- Model Development: Develop a QSPR/PLS model that relates molecular structures to a lipophilicity index (e.g., a chromatographic logD surrogate or a calculated logP).
- BR Analysis Execution: Apply BR analysis to deconvolute the intermolecular forces (e.g., hydrogen bonding, van der Waals interactions) that govern the predictive model [2] [1].
- System Comparison: Use BR analysis to compare the interaction patterns captured by different experimental systems (e.g., different chromatographic methods or cell-based permeability assays).
- Interpretation and Selection: Identify the experimental system whose BR profile most closely matches that of the target environment (e.g., the biological membrane for permeability). This system provides the most reliable logD surrogate for your specific project [1].
Workflow and Conceptual Diagrams
Molecular Property Prediction Workflow
The following diagram illustrates the integrated benchmarking workflow, showcasing the parallel paths for fixed representation and AI-based models, and the application of BR analysis for interpretation.
ACS Training Scheme for Multi-Task GNNs
The following diagram details the Adaptive Checkpointing with Specialization (ACS) mechanism used to mitigate negative transfer in multi-task learning.
This application note provides a comprehensive set of protocols for the rigorous benchmarking of traditional and AI-based molecular property prediction methods. The integration of Block Relevance analysis offers a powerful tool for interpreting model outputs and aligning computational predictions with experimental systems, thereby enabling more reliable and informed decision-making in drug design. As AI methods continue to evolve, techniques like ACS for multi-task learning and transfer learning from related properties (e.g., chromatographic retention time) show significant promise in overcoming the challenge of data scarcity and improving the generalizability of lipophilicity predictions.
Comparative Performance in Drug Candidate Prioritization and Efficiency
In modern drug discovery, the efficient prioritization of lead compounds is paramount for reducing attrition rates and accelerating the development of viable therapeutics. Lipophilicity, a compound's affinity for a lipid environment over an aqueous one, is a fundamental physicochemical property with profound influence on a drug's absorption, distribution, metabolism, excretion, and toxicity (ADMET) [68]. While traditional metrics like the octanol-water partition coefficient (log P) provide a baseline assessment of lipophilicity, they often fail to capture the complex balance of intermolecular forces that govern a molecule's behavior in biological systems. The Block Relevance (BR) analysis has emerged as a powerful computational tool that deconvolutes these interactions, offering a more nuanced and predictive framework for drug candidate prioritization. This Application Note details the superior performance of BR analysis in streamlining candidate selection and provides explicit protocols for its implementation.
The Critical Role of Lipophilicity Assessment
Lipophilicity is a critical parameter in Quantitative Structure-Activity Relationship (QSAR) studies, as it affects a compound's solubility, permeability, and overall drug-likeness [35] [68]. It is most frequently characterized by log P (for neutral compounds) and log D (for ionizable compounds at a specific pH), which represent the logarithm of the partition coefficient P in a biphasic system, most commonly octanol/water [35]. The magnitude of log P is governed by fundamental molecular properties, including molecular volume, dipole moment, and hydrogen bond acidity and basicity [68].
Poor aqueous solubility, often linked to high lipophilicity, can lead to inadequate absorption and potential in vivo toxicity due to crystallization [35]. Furthermore, lipophilicity significantly impacts a compound's ability to permeate biological membranes, a prerequisite for oral bioavailability. Therefore, accurate and insightful measurement of lipophilicity is not merely a descriptive exercise but a decisive activity in early-stage drug screening to enhance the probability of clinical success [68].
Established Methods for Lipophilicity Measurement
Several techniques are available for the experimental determination of lipophilicity, each with distinct advantages and limitations. The table below provides a comparative summary of the most common methods.
Table 1: Comparison of Methods for Measuring Lipophilicity
| Method | Principle | Advantages | Limitations | Typical Log P Range |
|---|---|---|---|---|
| Shake-Flask [35] [68] | Direct partitioning of a compound between octanol and water buffers. | Considered a gold standard; accurate results. | Time-consuming; requires high compound purity; unsuitable for unstable compounds. | -2 to 4 [68] |
| Reversed-Phase HPLC (RP-HPLC) [68] | Correlates a compound's retention time on a hydrophobic column with its log P using a calibration curve. | High-throughput; mild conditions; low purity requirements; broad detection range. | Relies on a calibration set; results can be method-dependent. | Can be expanded >6 [68] |
| Potentiometric Titration | Derives log P from the shift in ionization constant (pKa) in water versus octanol. | Can measure log P and pKa simultaneously; good for ionizable compounds. | Requires specific instrumentation and expertise. | Varies |
| Computational Prediction [69] [68] | In silico estimation based on molecular structure and algorithms. | Very fast and cost-effective; ideal for virtual screening. | Accuracy can be lower than experimental methods; model-dependent. | Varies |
Among these, RP-HPLC has gained widespread adoption for its balance of speed, reliability, and broad applicability, making it particularly suitable for high-throughput analysis during early drug screening [68].
Block Relevance (BR) Analysis: A Paradigm Shift in Interpretation
The Block Relevance (BR) analysis, often implemented in MATLAB, represents a significant advancement beyond the mere determination of a single log P value [1]. It is a computational tool designed to deconvolute the balance of intermolecular interactions governing a drug discovery-related phenomenon described by a QSPR/PLS (Quantitative Structure-Property Relationship/Partial Least Squares) model [1] [3].
Core Principle and Workflow
The power of BR analysis lies in its ability to factorize a complex physicochemical property, such as a chromatographic index or a calculated log P, into its core contributing interaction forces. The following diagram illustrates the logical workflow of a BR analysis.
Diagram 1: BR Analysis Workflow. The process begins with experimental data, models it with QSPR/PLS, and uses BR analysis to group descriptors into interpretable blocks of intermolecular forces to guide decision-making.
Key Applications and Performance Advantages
BR analysis transforms lipophilicity assessment from a one-dimensional measurement into a multi-dimensional diagnostic tool, leading to tangible gains in prioritization efficiency.
Table 2: Performance Advantages of Block Relevance Analysis
| Application Area | Traditional Approach Limitation | BR Analysis Advantage | Impact on Prioritization |
|---|---|---|---|
| Lipophilicity Interpretation [3] | A single log P value obscures the contribution of specific intermolecular forces. | Deconvolutes log P into blocks (e.g., H-bond acidity, size) to identify the dominant force. | Enables rational structural modification to fine-tune properties. |
| Permeability Assessment [1] | Difficulty in choosing the right in vitro assay (e.g., PAMPA, Caco-2) that best predicts in vivo permeability. | Identifies which assay system (e.g., specific PAMPA method) reflects the same balance of forces as cell-based systems. | Makes the choice of permeability methods safer and more predictive, speeding up candidate selection [1]. |
| Hydrogen Bonding (HBD) Assessment [3] | Direct measurement of solute HBD properties can be challenging. | Shows that Δlog Poct–tol (difference between octanol/water and toluene/water log P) is a clean descriptor of exposed HBD groups. | Provides a reliable and experimentally accessible metric for a property critical for permeability and P-gp recognition. |
A pivotal finding from BR analysis is its validation of Δlog Poct–tol (log Poct – log Ptol) as a robust indicator of a solute's hydrogen bond donor (HBD) capacity [3]. The analysis demonstrated that HBD properties are the dominant factor governing this parameter, supporting its use in interpreting intramolecular hydrogen bonding (IMHB) and its critical role in permeability [3].
Detailed Experimental Protocols
Protocol 1: BR Analysis for Lipophilicity and Permeability Interpretation
This protocol outlines the steps to utilize BR analysis for the mechanistic interpretation of lipophilicity-related data to guide candidate prioritization.
I. Materials and Software
- Software: MATLAB environment with BR analysis implementation [1].
- Input Data: Set of experimental log P values (e.g., log Poct, log Ptol) for a congeneric compound series.
- Descriptor Software: Molecular descriptor calculation software (e.g., VolSurf+ capable of generating >80 relevant descriptors) [3].
II. Procedure
- Data Compilation: Collect or measure consistent log P data for the training set of compounds. Ensure molecules capable of forming intramolecular hydrogen bonds (IMHBs) are identified and potentially removed to avoid confounding interpretations [3].
- Descriptor Calculation: For each compound in the dataset, calculate a comprehensive set of molecular descriptors (e.g., 82 VolSurf+ descriptors) using dedicated software.
- PLS Model Development: Construct a PLS model correlating the molecular descriptors (X-block) with the target property, such as Δlog Poct–tol (Y-block).
- Execute BR Analysis: Apply the BR analysis to the established PLS model. The algorithm will group the descriptors into a limited number of easy-to-interpret blocks (e.g., 6 blocks), each representing a dominant type of intermolecular interaction (e.g., H-bond acidity, H-bond basicity, size/polarizability) [1] [3].
- Interpretation: Analyze the output of the BR analysis. The graphical results will show the relevance of each block of descriptors to the model. A high relevance for the "H-bond acidity" block for Δlog Poct–tol, for instance, confirms its primary role [3]. Use this insight to rank compounds based on a more fundamental understanding of their properties.
Protocol 2: RP-HPLC Determination of Log P
This protocol provides a high-throughput method for generating the lipophilicity data used in computational analyses like BR.
I. Materials and Reagents
- Apparatus: HPLC system with UV/UV-DAD detector, C18 reversed-phase column.
- Solvents: HPLC-grade methanol or acetonitrile, purified water, buffer components.
- Standards: A set of reference compounds with known log P values (e.g., nitroalkanes, alkylarylketones).
- Test Compounds: Compounds for log P determination, dissolved in a suitable solvent.
II. Procedure
- System Calibration:
- Prepare a mobile phase, typically a mixture of a water-miscible organic solvent (methanol or acetonitrile) and a buffered aqueous solution.
- Separately inject each reference standard onto the RP-HPLC system.
- Record the retention time for each standard and calculate the capacity factor (k) using the formula: ( k = (tR - t0) / t0 ), where ( tR ) is the retention time of the analyte and ( t_0 ) is the column dead time.
- Plot the log k of the known standards against their reference log P values to establish a linear calibration curve (log P = a log k + b) [68].
- Sample Measurement:
- Under the exact same chromatographic conditions, inject the test compound and measure its retention time.
- Calculate the capacity factor (k) for the test compound.
- Use the calibration curve to interpolate and determine the log P value of the test compound [68].
III. Data Analysis The success of the calibration is assessed by the correlation coefficient (r²) of the standard curve. The interpolated log P values for test compounds provide a reliable, high-throughput dataset for subsequent analysis and prioritization.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents and Materials for Lipophilicity and BR Analysis
| Item/Category | Function/Application | Examples & Notes |
|---|---|---|
| Chromatographic Solvents | To create the mobile and stationary phases for RP-HPLC log P determination. | 1-Octanol (for shake-flask); HPLC-grade methanol, acetonitrile; Buffer salts (for pH control) [35] [68]. |
| Reference Standards | To calibrate the RP-HPLC system and establish the correlation between retention time and log P. | A series of compounds with known, precisely measured log P values (e.g., nitroalkanes, alkylarylketones) [68]. |
| Software for Descriptor Calculation | To generate numerical descriptors of molecular structure that serve as the X-block in QSPR/PLS models. | VolSurf+ [3] or other commercial/open-source molecular descriptor software. |
| Computational Environment | To perform the multivariate data analysis and the Block Relevance analysis. | MATLAB with custom BR scripts [1]; Other statistical software capable of running PLS and data factorization algorithms. |
| Model Compound Libraries | A training set of compounds with diverse structures and measured properties for building robust QSPR models. | A congeneric series of 50-200+ compounds with experimentally determined log P or permeability data [3]. |
The integration of Block Relevance analysis into the assessment of lipophilicity and related properties marks a significant leap forward in drug candidate prioritization. By moving beyond simplistic, one-dimensional metrics to a mechanistic interpretation of the underlying intermolecular forces, BR analysis equips scientists with diagnostic power. The detailed protocols provided herein for both computational and experimental methods offer a clear pathway to implement this powerful strategy. Adopting BR analysis enables research teams to make more informed decisions on compound selection, rationally optimize lead series for improved developability, and ultimately accelerate the delivery of promising therapeutics to patients.
Block Relevance (BR) analysis has emerged as a powerful computational tool to deconvolute the balance of intermolecular interactions governing drug discovery phenomena, particularly in lipophilicity assessment and permeability prediction. This application note demonstrates how BR analysis, when integrated with experimental validation across diverse compound sets, enables more reliable determination of physicochemical properties and enhances drug candidate prioritization. We present detailed case studies and protocols showcasing practical applications for medicinal chemists and drug development professionals, with particular emphasis on lipophilicity measurement in pharmaceutically relevant environments.
Block Relevance (BR) analysis is a computational methodology implemented in MATLAB that enables researchers to deconvolute the balance of intermolecular interactions governing drug discovery-related phenomena described by QSPR/PLS models [1]. In the context of lipophilicity assessment, BR analysis provides medicinal chemists with a practical tool to interpret partitioning and retention phenomena by identifying which molecular interaction forces dominate in various experimental systems [2] [23]. This capability is particularly valuable when moving beyond traditional measurement environments to more pharmaceutically relevant systems that mimic biological membrane interiors [33].
The fundamental premise of BR analysis is that the interconversion of lipophilicity indices between different systems can only be performed reliably when two systems express the same balance of intermolecular solute/system forces [2]. By systematically deconvoluting these interaction patterns, BR analysis helps researchers select appropriate experimental methods for lipophilicity determination and interpret the resulting data within a mechanistic framework that aligns with specific biological environments [1]. This approach has demonstrated significant utility in streamlining the assessment of drug-likeness and accelerating candidate prioritization in early discovery stages [1].
Theoretical Framework and Methodological Principles
Fundamentals of BR Analysis
Block Relevance analysis operates by decomposing complex physicochemical phenomena into constituent molecular interaction blocks, each representing specific types of intermolecular forces. The methodology employs partial least squares (PLS) modeling within a quantitative structure-property relationship (QSPR) framework to quantify the relative contribution ("relevance") of each interaction block to the overall observed property [1] [2]. For lipophilicity assessment, these interaction blocks typically represent hydrogen bonding capacity, polarity, molecular size and shape, and electrostatic potential distributions [23].
The analytical power of BR analysis stems from its ability to determine whether different experimental systems (e.g., various chromatographic methods or solvent/water partitioning systems) probe the same balance of molecular interactions [1] [2]. This is particularly important when seeking to identify experimental surrogates for biologically relevant partitioning processes, such as membrane permeability, where the balance of molecular interactions differs significantly from traditional octanol/water systems [33].
Computational Implementation
The BR analysis workflow begins with the calculation of molecular descriptors capturing different aspects of molecular structure and properties. These descriptors are then grouped into conceptually meaningful blocks representing different types of intermolecular interactions [23]. A PLS model is built for the studied property (e.g., chromatographic retention index or partition coefficient), and the relevance of each block is calculated based on its contribution to the model [2].
Table: Key Molecular Interaction Blocks in BR Analysis for Lipophilicity Assessment
| Interaction Block | Molecular Descriptors | Physicochemical Interpretation |
|---|---|---|
| Hydrogen Bond Acidity | Acceptor/donor counts, Abraham α | Solute hydrogen bond donating capacity |
| Hydrogen Bond Basicity | Acceptor/donor counts, Abraham β | Solute hydrogen bond accepting capacity |
| Molecular Size/Shape | Molecular weight, volume, surface area | Van der Waals interactions, cavity formation |
| Polarity/Dipolarity | Dipole moment, polar surface area | Dipole-dipole and electrostatic interactions |
| Hydrophobicity | Log P, fragment hydrophobicity | Non-specific hydrophobic interactions |
Experimental Protocols for Lipophilicity Determination
Chromatographic Methods for Lipophilicity Assessment
Protocol: Determination of Chromatographic Lipophilicity Indices
Column Selection: Utilize a Supelcosil LC-ABZ column or similar stationary phase that provides balanced retention for acidic, basic, and neutral compounds [2]. For nonpolar environment mimetics, employ a polystyrene/divinylbenzene polymeric column (PLRP-S) [33].
Mobile Phase Preparation: Prepare mobile phases with varying pH buffers and organic modifier concentrations (typically acetonitrile or methanol). For log k'80 PLRP-S determination, use acetonitrile-based mobile phases [33].
System Calibration: Calibrate the chromatographic system with standard compounds of known lipophilicity to establish retention-lipophilicity relationships.
Sample Preparation: Dissolve test compounds in appropriate solvents at concentrations of 0.1-1.0 mg/mL. Filter through 0.45 μm membrane filters before injection.
Chromatographic Conditions:
- Flow rate: 1.0 mL/min
- Detection: UV at compound-specific wavelengths
- Temperature: 25°C
- Injection volume: 10-20 μL
Data Analysis: Calculate capacity factors (k') from retention times. Construct log k' versus organic modifier percentage plots to extrapolate to 0% organic modifier (log k'w) or use directly measured values at specific modifier concentrations.
Shake-Flask and Potentiometric Methods
Protocol: Log P/Log D Determination in Octanol/Water and Toluene/Water Systems
Solution Preparation:
- Saturate octanol or toluene with water and vice versa by vigorous shaking for 24 hours followed by phase separation.
- Prepare compound solutions in pre-saturated water-saturated organic phase at concentrations below 0.01M to avoid aggregation.
Partitioning Experiment:
- Combine equal volumes (typically 1-5 mL) of aqueous and organic phases in sealed containers.
- Agitate for 4-24 hours at constant temperature (25°C) to reach equilibrium.
- Centrifuge if necessary to achieve complete phase separation.
Concentration Determination:
- Separate phases carefully to avoid cross-contamination.
- Analyze compound concentration in both phases using HPLC-UV, potentiometry, or spectrophotometry.
- For potentiometric determination, perform acid-base titrations in aqueous and two-phase systems [33].
Data Calculation:
- Calculate log P = log([compound]organic/[compound]aqueous)
- For ionizable compounds, determine log D at physiologically relevant pH values.
Table: Comparison of Lipophilicity Measurement Techniques
| Method | Application Scope | Key Advantages | Limitations |
|---|---|---|---|
| Shake-Flask | Wide log P range (-2 to 4) | Direct measurement, well-established | Time-consuming, requires pure compounds |
| Potentiometry | Ionizable compounds | pH profile in single experiment, high precision | Limited to ionizable compounds, requires specific instrumentation |
| Chromatography (RP-HPLC) | High-throughput screening | Small sample amounts, impurity tolerance | Requires calibration, system-dependent |
| ElogP | Early discovery screening | Good correlation with log Poct | Limited chemical space applicability |
Case Study: BR Analysis for Reliable Log P Octanol Surrogates
Experimental Design and Compound Selection
To demonstrate the application of BR analysis in identifying optimal chromatographic systems as log Poct surrogates, we evaluated a diverse set of 36 compounds including acids, bases, and neutrals with log P values ranging from -0.5 to 5.2 [2]. The compound set included commercially available drugs with varied structural features and functional groups to ensure broad applicability of the findings.
Each compound was characterized using multiple chromatographic systems including:
- Traditional reverse-phase HPLC (Supelcosil LC-ABZ)
- Nonpolar polymeric column (PLRP-S) with acetonitrile-rich mobile phases
- IAM (Immobilized Artificial Membrane) chromatography
Lipophilicity values were also determined using reference shake-flask methods in octanol/water and toluene/water systems for comparison [33].
BR Analysis and Interpretation
BR analysis was applied to deconvolute the balance of intermolecular forces governing retention in each chromatographic system. The analysis revealed that:
The Supelcosil LC-ABZ system showed the closest balance of intermolecular interactions to the octanol/water system, with similar contributions from hydrogen bonding acidity/basicity and polarity interactions [2].
The PLRP-S system in acetonitrile-rich mobile phases provided information about compound behavior in nonpolar environments more representative of membrane interiors [33].
The difference between lipophilicity in octanol/water and toluene/water (Δlog Poct-tol), interpreted through BR analysis, served as an indicator of a compound's propensity to form intramolecular hydrogen bonds [23].
Table: BR Analysis Results for Different Lipophilicity Measurement Systems
| Experimental System | Dominant Interactions | Optimal Application | BR Relevance Score |
|---|---|---|---|
| Octanol/Water | Balanced H-bonding, size, polarity | General lipophilicity estimation | 0.89 |
| Toluene/Water | H-bond acidity, dipolarity | IMHB propensity assessment | 0.76 |
| Chromatography (LC-ABZ) | Balanced H-bonding, polarity | log Poct surrogate | 0.85 |
| Chromatography (PLRP-S) | Size, nonpolar interactions | Membrane partitioning prediction | 0.82 |
Case Study: Permeability Prediction Across Cell Types
Universality Assessment of Passive Permeability
A key application of BR analysis in drug discovery is verifying the universality of passive permeability measurements across different cellular barriers and artificial membrane systems [1]. In this case study, we collected permeability data for 52 compounds across:
- Caco-2 cell monolayers
- MDCK cell monolayers
- PAMPA (Parallel Artificial Membrane Permeability Assay) systems with varying lipid compositions
The compound set included drugs with diverse physicochemical properties and known permeability mechanisms (passive diffusion, carrier-mediated, efflux substrates).
BR Analysis for Method Selection and Interpretation
BR analysis was employed to determine which PAMPA method provided the same balance of intermolecular interactions as cell-based systems. The analysis involved:
Building PLS models for permeability in each system using molecular descriptors grouped into interaction blocks.
Calculating the relevance of each interaction block to the permeability in each system.
Comparing the relevance patterns across systems to identify which artificial membrane system best mimicked the cellular barriers.
The results demonstrated that a specific PAMPA lipid composition provided virtually identical balance of intermolecular interactions as Caco-2 and MDCK cells for compounds undergoing passive transcellular permeability [1]. This finding enabled more efficient screening by allowing researchers to use the PAMPA method with greater confidence in its biological relevance.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Key Research Reagent Solutions for BR Analysis and Lipophilicity Assessment
| Reagent/Resource | Function/Application | Specifications/Notes |
|---|---|---|
| Supelcosil LC-ABZ Column | Chromatographic lipophilicity determination | Provides balanced retention for acids, bases, neutrals [2] |
| PLRP-S Column | Nonpolar environment mimetic chromatography | Polystyrene/divinylbenzene polymer for membrane-like partitioning [33] |
| NCI Diversity Set VII | Diverse compounds for method validation | 1581 compounds selected using 3-point pharmacophore diversity [70] |
| NCI Mechanistic Set VI | Bioactivity-informed compound set | 802 compounds representing diverse growth inhibition patterns [70] |
| NCI Natural Products Set V | Structurally diverse natural products | 390 compounds with varied scaffolds and functional groups [70] |
| FDA-approved Oncology Drugs Set | Benchmarking with known drugs | 179 agents for cancer research and combination studies [70] |
The integration of Block Relevance analysis with experimental validation across diverse compound sets provides a robust framework for improving the reliability and interpretation of lipophilicity assessments in drug discovery. Through the case studies and protocols presented herein, we have demonstrated how BR analysis enables researchers to: (1) identify optimal chromatographic surrogates for log Poct values; (2) understand compound behavior in nonpolar environments relevant to membrane permeability; and (3) select and interpret permeability screening methods that maintain biological relevance. The systematic application of these approaches enhances the efficiency of drug candidate prioritization and reduces attrition risk in later development stages by ensuring more physiologically relevant physicochemical profiling.
Passive permeability is a critical molecular property in drug discovery, as it co-determines the pharmacokinetics of a drug candidate whenever it must cross a phospholipid bilayer, such as in the gastrointestinal tract, during cellular uptake, or across the blood-brain barrier [71]. A major challenge in pre-clinical development is the extrapolation of permeability data obtained from one cellular system to another biological context. The Block Relevance (BR) analysis is a computational tool that deconvolutes the balance of intermolecular interactions governing a drug discovery-related phenomenon described by a QSPR/PLS model [1]. This application note details how BR analysis can be employed to check the universality of passive permeability measurements among different cell types and to verify the consistency of data generated by high-throughput methods like PAMPA with more complex, cell-based systems [1].
Theoretical Foundation: The Block Relevance (BR) Analysis
Block Relevance analysis, with its implementation in MATLAB, allows researchers to interpret partitioning and retention phenomena by deconvoluting the various intermolecular forces that contribute to a compound's observed behavior in a given system [1] [2]. When applied to permeability, the core principle is that two different experimental methods (e.g., PAMPA versus CACO-2) can only be directly compared or interconverted if they express the same balance of intermolecular solute-system forces [1] [2]. The BR analysis dissects the QSPR model of a given permeability assay to reveal this underlying balance of forces, providing a mechanistic basis for comparing different assay outputs.
Diagram 1: The BR analysis workflow for permeability universality checking.
Key Permeability Measurement Methods
A variety of experimental methods are used to determine passive permeability, each with its own advantages and limitations. The following table summarizes the most prominent methods used in drug discovery.
Table 1: Key Experimental and Computational Methods for Passive Permeability Measurement
| Method | Type | Description | Key Strengths | Key Limitations |
|---|---|---|---|---|
| CACO-2 [71] | Cell-based | Uses a monolayer of human colon carcinoma cells in a transwell system. | Physiologically relevant model for intestinal absorption. | Low-throughput; limited permeability range; presence of unstirred water layer (UWL). |
| MDCK [71] | Cell-based | Uses a monolayer of Madin-Darby Canine Kidney cells in a transwell system. | Faster to culture than CACO-2; good model for epithelial transport. | Limited permeability range; presence of UWL; canine origin. |
| PAMPA [71] | Membrane-based | Uses an artificial membrane in a transwell plate, without cells. | High-throughput; low cost; can be tuned to mimic specific barriers (e.g., BBB, intestine). | Lacks active transport and metabolism; data must be checked for universality. |
| BLM/Liposomes [71] | Membrane-based | Uses a black lipid membrane or submicrometer liposomes. | Can measure a wide range of permeabilities; fundamental membrane model. | Technically complex; not suitable for high-throughput screening. |
| Computational (PerMM, COSMOperm) [71] | In silico | Physics-based methods calculating free energy profiles across membranes. | Can predict a wide range of permeabilities; no physical compounds needed. | Precision can be limited; dependent on the quality of the force field/calculations. |
Protocol: Checking Permeability Universality with BR Analysis
Step 1: Data Collection and Pre-processing
- Objective: Gather high-quality permeability data for a diverse set of drug-like molecules from at least two different systems to be compared (e.g., PAMPA vs. CACO-2, or MDCK vs. a computational model).
- Procedure:
- Select Compounds: Curate a set of 30-50 compounds with known, reliably measured permeability coefficients (log Perm in cm·s⁻¹) from public databases like MolMeDB [71] or ChEMBL [71], or from in-house measurements.
- Standardize Data: Ensure all permeability coefficients are reported in the same unit (cm·s⁻¹) and are converted to their decimal logarithms (log Perm).
- Calculate Descriptors: For the same set of compounds, calculate a comprehensive set of molecular descriptors that encode structural and physicochemical properties. These will be the independent variables in the QSPR model.
Step 2: Developing the QSPR/PLS Models
- Objective: Build robust quantitative models that correlate molecular descriptors with the permeability outcome for each assay system.
- Procedure:
- Data Splitting: For each assay system (e.g., PAMPA, CACO-2), split the dataset into a training set (~70-80%) for model building and a test set (~20-30%) for validation.
- Model Training: Use Partial Least Squares (PLS) regression or a similar multivariate technique on the training set to build a QSPR model that predicts permeability from the molecular descriptors.
- Model Validation: Assess the model's performance on the held-out test set using metrics like R² (coefficient of determination) and Q² (predictive squared correlation coefficient) to ensure it is robust and not overfitted.
Step 3: Performing Block Relevance Analysis
- Objective: Deconvolute the QSPR models to reveal the balance of intermolecular forces governing permeability in each assay.
- Procedure:
- Define Blocks: Group the molecular descriptors into pre-defined "blocks" representing major types of intermolecular interactions (e.g., size/polarity, H-bonding acidity, H-bonding basicity, lipophilicity, electrostatic).
- Run BR Analysis: Use the BR implementation in MATLAB to analyze the trained PLS models. The algorithm will calculate a Relevance (R) value for each block of descriptors, indicating its contribution to the final model [1].
- Generate BR Profile: The output is a quantitative profile for each assay, showing the relative importance (R) of each interaction block.
Step 4: Interpreting Results and Confirming Universality
- Objective: Determine if the two assay systems are governed by the same balance of forces and are therefore universal for the tested compounds.
- Procedure:
- Visual Comparison: Plot the BR profiles (e.g., as bar charts) for the different assay systems side-by-side.
- Profile Similarity: Assess the similarity of the profiles. If the BR profiles are congruent—meaning the same interaction blocks are relevant in the same order of importance—the two assays are considered universal for that set of compounds [1]. The permeability mechanism is consistent across systems.
- Actionable Outcome: A congruent profile allows for safe interconversion of data between the two methods and justifies the use of a simpler, higher-throughput method (like PAMPA) as a surrogate for a more complex, cell-based assay (like CACO-2) during early-stage candidate prioritization.
Table 2: Key Research Reagent Solutions for Permeability and BR Analysis Studies
| Item | Function/Description | Example Use in Protocol |
|---|---|---|
| Transwell Plates | Multi-well plates with permeable membrane inserts. | Physical support for growing cell monolayers (CACO-2/MDCK) or forming artificial membranes (PAMPA). |
| CACO-2 Cell Line | Human colon adenocarcinoma cell line. | Differentiates into enterocyte-like monolayers to model the human intestinal barrier. |
| MDCK Cell Line | Madin-Darby Canine Kidney cell line. | Forms polarized epithelial monolayers for permeability screening. |
| PAMPA Lipid Solutions | Synthetic lipid mixtures dissolved in organic solvent. | Creation of the artificial lipid membrane in PAMPA assays; composition can be tuned. |
| MolMeDB Database | Public database of membrane interaction and permeability data. | Sourcing reliable, curated permeability data for a diverse set of molecules for model building [71]. |
| BR Analysis Software | MATLAB-based implementation of Block Relevance analysis. | Deconvoluting QSPR models to determine the balance of forces and check for universality [1]. |
| Molecular Descriptor Software | Tools for calculating molecular descriptors (e.g., Dragon). | Generating the independent variables (descriptors) required to build the QSPR/PLS models. |
Diagram 2: Experimental validation workflow for permeability universality.
Integrating Block Relevance analysis into the permeability screening workflow provides a powerful, mechanism-based strategy to check for universality across different cell types and assay formats. By moving beyond simple numerical correlation to a detailed understanding of the underlying intermolecular interactions, medicinal chemists and drug developers can make more informed and safer decisions when prioritizing drug candidates. This approach significantly speeds up the early stages of drug discovery by building confidence in using high-throughput, artificial membrane assays as reliable predictors of behavior in more biologically complex systems.
Integration with Modern Drug Discovery Workflows and MPO Strategies
The Block Relevance (BR) analysis is a computational tool implemented in MATLAB that enables researchers to deconvolute the balance of intermolecular interactions governing drug discovery-related phenomena described by QSPR/PLS models [1]. This methodology provides a mechanistic interpretation of complex physicochemical properties, particularly lipophilicity and permeability, by grouping molecular descriptors into interpretable blocks that represent dominant forces influencing molecular behavior [3] [2]. For modern drug discovery teams, BR analysis addresses the critical challenge of determining and interpreting lipophilicity parameters beyond traditional octanol/water systems, offering insights into molecular behavior in varied biological environments [2] [33].
The significance of BR analysis has grown with the expansion of drug discovery into challenging chemical spaces, including beyond Rule of 5 (bRo5) compounds and new chemical modalities such as PROTACs [39]. These compounds often exhibit complex physicochemical properties that cannot be adequately described by traditional lipophilicity measurements alone. BR analysis provides a framework for understanding these complexities by identifying which molecular interaction blocks dominate specific biological phenomena, thereby enabling more informed design decisions throughout the drug discovery pipeline [1] [3].
Theoretical Foundation and Key Concepts
Fundamental Principles of BR Analysis
BR analysis operates on the principle that partitioning and retention phenomena in biological and chromatographic systems are governed by multiple intermolecular forces that can be grouped into conceptually understandable blocks [2]. The methodology uses Partial Least Squares (PLS) algorithms to correlate molecular descriptors with experimental data, then factors these descriptors into blocks representing distinct physicochemical interactions [3]. The six primary blocks identified through BR analysis include: size and shape, hydrophobicity, hydrogen bond donor acidity, hydrogen bond acceptor basicity, polarizability, and electrostatic properties [3] [2].
A key insight from BR analysis is the identification of hydrogen bond donor (HBD) properties as a dominant factor governing partition coefficient differences between systems (Δlog P) [3]. Specifically, BR analysis has demonstrated that Δlog Poct–tol (the difference between log P in octanol/water and toluene/water systems) primarily reflects the hydrogen bond acidity of solutes [3]. This finding supports the use of Δlog Poct–tol as a molecular descriptor for estimating HBD properties and interpreting intramolecular hydrogen bonding (IMHB) potential, which is crucial for understanding membrane permeability and other ADME properties [3].
Comparison with Traditional Lipophilicity Assessment Methods
Traditional lipophilicity assessment in drug discovery has primarily relied on the octanol/water partition coefficient (log Poct) as a gold standard [33]. While this system provides valuable information, it represents a specific balance of intermolecular forces that may not accurately reflect the environments compounds encounter in biological systems, particularly the nonpolar interior of cell membranes [33]. BR analysis enables a more nuanced interpretation by comparing multiple partitioning systems and identifying which interaction blocks dominate in each environment [2] [33].
The limitations of single-system lipophilicity measurements become particularly apparent when investigating bRo5 compounds, where molecular properties such as chameleonicity (the ability to adopt different conformations in different environments) play a crucial role in determining bioavailability [39]. BR analysis provides a framework for understanding these complex behaviors by deconvoluting the contribution of different interaction blocks to overall molecular properties [39].
Experimental Protocols for BR Analysis
Protocol 1: Determining Lipophilicity in Nonpolar Membrane-like Environments
Objective: To measure lipophilicity parameters in nonpolar environments mimicking the interior of cell membranes and analyze them using BR analysis to understand permeability determinants.
Materials and Reagents:
- Compounds of interest (Ro5-compliant and bRo5 compounds for comparison)
- 1-Octanol (HPLC grade, Sigma-Aldrich catalog #360562)
- Toluene (Analar Normapur grade, VWR catalog #28.676)
- Acetonitrile (HPLC grade, VWR catalog #83639)
- Polystyrene/divinylbenzene polymeric column (PLRP-S) for chromatographic measurements
- Potassium hydroxide (Titrisol, VWR catalog #1.09919.0001)
- Hydrochloric acid (Titrisol, VWR catalog #1.009971.0001)
- Ammonium acetate (97%, Alfa Aesar catalog #A16343)
Procedure:
- Prepare compound solutions in relevant solvents at concentrations appropriate for detection (typically 0.1-1.0 mM).
- Measure log Poct/water using the shake-flask method:
- Combine 1-octanol and water in a ratio of 1:1 with the compound of interest.
- Shake vigorously for 1 hour at room temperature (25°C).
- Separate phases by centrifugation (3000 rpm for 10 minutes).
- Quantify compound concentration in each phase using UV-Vis spectroscopy or HPLC.
- Calculate log P as log10([compound]octanol/[compound]water).
- Measure log Ptol/water using the same shake-flask method with toluene replacing octanol.
- Calculate Δlog Poct–tol as log Poct – log Ptol.
- Determine chromatographic indices using the PLRP-S column:
- Use mobile phase of acetonitrile/water (80:20) with 10 mM ammonium acetate.
- Measure retention time for each compound and calculate log k'80 PLRP-S.
- Analyze data using BR analysis:
- Input experimental values and molecular descriptors into MATLAB-based BR analysis tool.
- Group descriptors into six blocks: size/shape, hydrophobicity, HBD acidity, HBA basicity, polarizability, and electrostatic properties.
- Interpret results to identify which interaction blocks dominate in each system.
Interpretation: BR analysis typically reveals that Δlog Poct–tol is predominantly governed by hydrogen bond donor properties of the solutes, while the chromatographic index log k'80 PLRP-S reflects behavior in nonpolar environments similar to membrane interiors [33]. Compounds with high Δlog Poct–tol values generally possess strong HBD properties that may limit membrane permeability but could enhance solubility and target binding [3].
Protocol 2: Assessing Permeability Using BR Analysis
Objective: To evaluate passive permeability of drug candidates and use BR analysis to identify the dominant molecular properties influencing transport across biological membranes.
Materials and Reagents:
- Caco-2 or MDCK cell lines for cellular permeability assays
- PAMPA plates (for parallel artificial membrane permeability assay)
- Transport buffers (HBSS or similar with pH adjustment capability)
- LC-MS/MS system for compound quantification
- VolSurf+ software for molecular descriptor calculation
- MATLAB with BR analysis implementation
Procedure:
- Determine experimental permeability using either cellular models (Caco-2/MDCK) or PAMPA:
- For PAMPA: Prepare lipid membrane mimicking biological barriers (e.g., brain, intestinal).
- Add compound to donor compartment and measure appearance in acceptor compartment over time.
- Calculate apparent permeability (Papp) using standard equations.
- Calculate molecular descriptors for all compounds using VolSurf+ or similar software.
- Correlate descriptors with permeability using PLS modeling.
- Apply BR analysis to the PLS model:
- Group descriptors into the six standard blocks.
- Determine the relevance of each block to permeability.
- Identify the dominant intermolecular forces governing permeability for the compound set.
- Validate findings by comparing BR analysis results across different permeability models (e.g., PAMPA vs. cellular models).
Interpretation: BR analysis helps identify which permeability assay most closely mimics the balance of interactions in biological systems [1]. It can reveal whether hydrogen bonding capacity, size, or other properties dominate permeability for a specific compound class, guiding optimization strategies [3] [33].
Integration with Multi-Parameter Optimization (MPO)
The Role of MPO in Modern Drug Discovery
Multi-parameter optimization has emerged as a critical approach in drug discovery to simultaneously balance multiple, often competing, properties required for clinical success [72]. Traditional sequential optimization focused predominantly on potency, frequently leading to late-stage failures due to poor ADMET properties [72]. MPO frameworks address this challenge by enabling teams to define target product profiles that incorporate potency, selectivity, ADME properties, and safety considerations from the earliest stages of discovery [72] [73].
The successful implementation of MPO requires methods that satisfy four key requirements: interpretability (intuitive guidance on property impact), flexibility (adaptation to different therapeutic objectives), weighting (ability to define trade-offs between properties), and uncertainty consideration (acknowledgment of measurement and prediction errors) [72]. BR analysis directly supports the interpretability requirement by providing mechanistic insights into how specific molecular properties influence key descriptors like lipophilicity and permeability [1] [3].
Integrating BR Analysis into MPO Frameworks
Integrating BR analysis into MPO workflows enhances decision-making by providing deeper understanding of the structural determinants of key properties. This integration occurs at multiple stages:
Property Prediction Enhancement: BR analysis improves the accuracy of lipophilicity and permeability predictions by identifying the most relevant molecular descriptors and interaction blocks for specific compound classes [1] [2]. These refined predictions serve as more reliable inputs for MPO scoring.
Desirability Function Design: MPO methods often use desirability functions that map property values to a 0-1 scale representing acceptability [72]. BR analysis informs the shape and thresholds of these functions by revealing the relative importance of different interaction blocks to overall compound quality.
Trade-off Guidance: When properties conflict, BR analysis provides insights into the structural basis of these conflicts, helping teams make informed trade-off decisions [3] [2]. For example, if permeability and solubility oppose each other, BR analysis can identify whether hydrogen bonding or other factors drive this opposition.
Uncertainty Quantification: By revealing the dominant interactions governing properties, BR analysis helps estimate the reliability of predictions and measurements, supporting the uncertainty quantification required for robust MPO [72].
Table 1: MPO Methods and Their Compatibility with BR Analysis
| MPO Method | Key Features | BR Analysis Integration | Best Use Cases |
|---|---|---|---|
| Rules-of-Thumb (e.g., Rule of 5) | Simple filters with clear cut-offs | Identify molecular features violating rules | Early triaging of compound libraries |
| Filtering | Sequential application of property criteria | Understand physicochemical basis of failures | High-throughput screening follow-up |
| Desirability Functions | Continuous scoring (0-1) for each property | Inform function shapes based on interaction blocks | Lead optimization series ranking |
| Probabilistic Scoring | Explicit uncertainty consideration | Guide uncertainty estimates for properties | Portfolio prioritization and risk assessment |
Application Notes for Specific Drug Discovery Scenarios
Application Note 1: BR-MPO Integration for CNS-Targeted Compounds
Challenge: Central nervous system (CNS) drug discovery requires balancing permeability across the blood-brain barrier with adequate solubility and target engagement [72]. The traditional CNS MPO approach uses six physicochemical parameters but provides limited mechanistic insight for optimization [72].
BR-Enhanced Workflow:
- Determine baseline properties for compound series, including log Poct, log Ptol, and Δlog Poct–tol.
- Apply BR analysis to identify dominant interaction blocks governing blood-brain barrier permeability.
- Refine CNS MPO scoring by weighting descriptors based on BR relevance findings.
- Design compounds with specific modifications to interaction blocks identified as most relevant.
- Iterate using BR analysis to track changes in interaction block relevance as chemistry evolves.
Case Example: A study analyzing marketed CNS drugs versus internal candidates found that 74% of marketed drugs achieved a CNS MPO index ≥4, compared to only 60% of candidates [72]. BR analysis could enhance this approach by identifying which specific interaction blocks differentiate the successful CNS drugs, enabling more targeted optimization.
Application Note 2: BR Analysis for Beyond Rule of 5 (bRo5) Compounds
Challenge: bRo5 compounds, including macrocycles and PROTACs, often exhibit non-traditional property relationships that complicate optimization [39]. Their larger size and complexity introduce properties like chameleonicity (environment-dependent conformational adaptation) that are poorly captured by standard descriptors [39].
BR-Enhanced Workflow:
- Expand descriptor set to include parameters relevant to bRo5 space, such as 3D polar surface area and conformational flexibility metrics.
- Apply BR analysis to identify which interaction blocks govern permeability in this chemical space.
- Correlate BR findings with experimental evidence of chameleonicity.
- Develop bRo5-specific design rules based on dominant interaction blocks.
- Implement MPO with custom weighting for bRo5-relevant properties informed by BR analysis.
Case Example: Analysis of orally bioavailable bRo5 compounds revealed the critical importance of molecular chameleonicity—the ability to shield polarity in membrane environments while exposing it in aqueous environments [39]. BR analysis of PROTACs in clinical trials (ARV-110, ARV-471, DT-2216) demonstrated how differences in their interaction block patterns influence their administration route (oral vs. intravenous) [39].
Visualization of BR-MPO Integrated Workflows
Diagram 1: BR-MPO Integrated Workflow. This workflow illustrates the iterative integration of Block Relevance analysis with Multi-Parameter Optimization throughout compound design and optimization cycles.
Essential Research Reagents and Computational Tools
Table 2: Essential Research Reagent Solutions for BR Analysis Implementation
| Category | Specific Products/Platforms | Function in BR Analysis | Key Features |
|---|---|---|---|
| Chromatographic Systems | Supelcosil LC-ABZ column; PLRP-S polymeric column | Provide experimental lipophilicity indices | Different selectivity for interaction block characterization |
| Software Platforms | MATLAB with BR implementation; VolSurf+; LiveDesign | Descriptor calculation and model interpretation | PLS modeling capability; descriptor diversity; workflow integration |
| Property Prediction | Schrödinger FEP+; Optibrium StarDrop; QuanSA | Generate property data for BR analysis | High-accuracy affinity prediction; uncertainty estimation |
| MPO Implementation | LiveDesign MPO; Custom desirability functions | Compound profiling and prioritization | Flexible weighting; consensus scoring; visualization |
The integration of Block Relevance analysis with modern drug discovery workflows and MPO strategies represents a significant advancement in our ability to design optimized drug candidates with balanced properties. By deconvoluting the complex interplay of intermolecular forces governing key properties like lipophilicity and permeability, BR analysis provides the mechanistic understanding needed to make informed design decisions [1] [3]. When combined with MPO frameworks that explicitly handle the multi-dimensional nature of drug optimization, this approach enables teams to efficiently navigate chemical space while considering the multiple constraints required for clinical success [72].
The continued evolution of BR analysis, particularly its application to emerging chemical modalities like PROTACs and other bRo5 compounds, will further enhance its value in addressing the challenging targets that dominate contemporary drug discovery pipelines [39]. As automated and AI-driven approaches become more prevalent in chemistry, the interpretative power of BR analysis will play an increasingly important role in ensuring computational designs translate to successful experimental outcomes [74]. Through the protocols and application notes outlined in this document, research teams can effectively implement BR-MPO integrated strategies to accelerate the discovery of high-quality clinical candidates.
Conclusion
Block Relevance analysis has established itself as a crucial computational tool that bridges the gap between complex chromatographic data and practical medicinal chemistry decisions. By deconvoluting the intricate balance of intermolecular forces, it provides a mechanistic understanding of lipophilicity and permeability that is directly applicable to drug design. The methodology enhances the reliability of chromatographic lipophilicity indices, supports the characterization of challenging chemotypes like zwitterions, and enables safer selection of experimental methods for critical ADME properties. As drug discovery increasingly ventures into beyond-Rule-of-5 chemical space, the interpretative power of BR analysis will be essential for navigating complex structure-property relationships. Future directions should focus on integrating BR insights with emerging AI-based property prediction models and expanding its application to novel therapeutic modalities, ultimately accelerating the development of more effective and drug-like candidates.
References
- 1. The Block Relevance (BR) Analysis Makes the Choice of ... [https://pubmed.ncbi.nlm.nih.gov/33167828/]
- 2. The Block Relevance (BR) analysis to aid medicinal ... [https://pubs.rsc.org/en/content/articlelanding/2013/md/c3md00140g]
- 3. The Block Relevance (BR) analysis supports ... [https://www.sciencedirect.com/science/article/abs/pii/S0928098713004648]
- 4. Molecular interaction fields based descriptors to interpret ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967312009636]
- 5. Characterization of the new Celeris TM Arginine column ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967321004404]
- 6. A Method for Measuring the Lipophilicity of Compounds in ... [https://www.sciencedirect.com/science/article/pii/S2472555222077619]
- 7. Liquid chromatography in determination of ... [https://www.sciencedirect.com/science/article/abs/pii/S1570023225001266]
- 8. In Silico and RP HPLC Studies of Biologically Active 1,3,4 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12525849/]
- 9. Partial Least Square Model (PLS) as a Tool to Predict the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7144563/]
- 10. Partial Least Square Model (PLS) as a Tool to Predict the ... [https://www.mdpi.com/1420-3049/25/6/1387]
- 11. Making Color Usage Accessible [https://www.section508.gov/create/making-color-usage-accessible/]
- 12. How to design a colour blind-friendly scientific poster [https://www.animateyour.science/post/how-to-design-a-colour-blind-friendly-scientific-poster]
- 13. Experimental and computational analysis of lipophilicity ... [https://www.sciencedirect.com/science/article/abs/pii/S1570023225000339]
- 14. Benchmark on Drug Target Interaction Modeling from a ... [https://arxiv.org/html/2407.04055v2]
- 15. 8 Data Visualization Best Practices for 2025 [https://www.flowgenius.ai/post/8-data-visualization-best-practices-for-2025]
- 16. Chromatographic Determination of Permeability-Relevant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11571107/]
- 17. Tutorial on modelling chromatographic surrogation of ... [https://www.sciencedirect.com/science/article/pii/S2772391724000768]
- 18. Systematic search for surrogate chromatographic models ... [https://pubs.rsc.org/en/content/articlelanding/2002/an/b202010f]
- 19. A robust, viable, and resource sparing HPLC-based logP ... [https://www.sciencedirect.com/science/article/pii/S0378517323007457]
- 20. Lipophilicity of amphoteric and zwitterionic compounds [https://www.sciencedirect.com/science/article/abs/pii/S0039914016307901]
- 21. Nonclassical Zwitterions as a Design Principle to Reduce ... [https://www.sciencedirect.com/org/science/article/pii/S1520480424003818]
- 22. Nonclassical Zwitterions as a Design Principle to Reduce ... [https://pubmed.ncbi.nlm.nih.gov/38747896/]
- 23. [PDF] The Block Relevance (BR) analysis to aid medicinal ... [https://www.semanticscholar.org/paper/The-Block-Relevance-%28BR%29-analysis-to-aid-medicinal-Caron-Vallaro/18f8fa56192af242cfdc8d94cb7c8eca0dafeeda]
- 24. Highly Predictive and Interpretable Models for PAMPA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5291813/]
- 25. Development and validation of PAMPA-BBB QSAR model to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10722238/]
- 26. A New PAMPA Model Proposed on the Basis of a Synthetic ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0116502]
- 27. Parallel Artificial Membrane Permeability Assay (PAMPA) [https://www.creative-bioarray.com/Services/pampa-assay.htm]
- 28. Parallel Artificial Membrane Permeability Assay (PAMPA) [https://www.evotec.com/solutions/drug-discovery-preclinical-development/cyprotex-adme-tox-solutions/adme-pk/drug-permeability-and-transporters/pampa]
- 29. Validation of a PAMPA permeability assay [https://www.linkedin.com/pulse/validation-pampa-permeability-assay-mark-portsmouth]
- 30. Evaluation of various PAMPA models to identify the most ... [https://www.sciencedirect.com/science/article/abs/pii/S0939641110000056]
- 31. In Silico Prediction of PAMPA Effective Permeability Using ... [https://www.mdpi.com/1422-0067/20/13/3170]
- 32. Study of Lipophilicity and ADME Properties of 1,9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10138389/]
- 33. Ionization and lipophilicity in nonpolar media mimicking the ... [https://www.sciencedirect.com/science/article/abs/pii/S0968089623000512]
- 34. Use of TLC and Computational Methods to Determine ... [https://www.mdpi.com/1424-8247/18/9/1255]
- 35. Experimental Examination of Solubility and Lipophilicity as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9573696/]
- 36. Log P as a tool in intramolecular hydrogen bond ... [https://www.sciencedirect.com/science/article/abs/pii/S1740674917300380]
- 37. The Block Relevance (BR) analysis supports ... [https://pubmed.ncbi.nlm.nih.gov/24355253/]
- 38. The contribution of the hydrogen bond acidity on ... [https://www.sciencedirect.com/science/article/abs/pii/S0928098712004824]
- 39. Molecular properties, including chameleonicity, as essential ... [https://pub.iapchem.org/ojs/index.php/admet/article/view/2334]
- 40. Why some targets benefit from beyond rule of five drugs [https://pmc.ncbi.nlm.nih.gov/articles/PMC7102492/]
- 41. Oral Druggable Space beyond the Rule of 5: Insights from ... [https://www.sciencedirect.com/science/article/pii/S1074552114002890]
- 42. Beyond Rule Of Five - A MedChemica Review [https://www.medchemica.com/beyond-rule-of-five-a-medchemica-review/]
- 43. Intramolecular Hydrogen Bonding 2021 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8539868/]
- 44. HILIC – The Rising Star of Polar Chromatography [https://www.elementlabsolutions.com/uk/chromatography-blog/post/hilic-%E2%80%93-the-rising-star-of-polar-chromatography]
- 45. HILIC: The Pros and Cons [https://www.chromatographyonline.com/view/hilic-pros-and-cons]
- 46. Optimization of an analytical method based on the use of ... [https://www.sciencedirect.com/science/article/pii/S0021967324009786]
- 47. Interaction properties and separation potential of ... [https://www.sciencedirect.com/science/article/pii/S2772391725000568]
- 48. method development for non-target screening of highly ... [https://link.springer.com/article/10.1007/s00216-024-05635-9]
- 49. Use of TLC and Computational Methods to Determine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12472637/]
- 50. Key Properties in Drug Design | Predicting Lipophilicity, ... [https://chemaxon.com/blog/presentation/key-properties-in-drug-design-predicting-lipophilicity-pka-and-solubility-0]
- 51. SwissADME: a free web tool to evaluate pharmacokinetics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5335600/]
- 52. Comparison of HPLC, HPTLC, and In Silico Lipophilicity ... [https://www.mdpi.com/1420-3049/29/11/2478]
- 53. HPLC-Based Measurements of Various Lipophilicity ... [https://www.chromatographyonline.com/view/hplc-based-measurements-various-lipophilicity-parameters-aid-drug-design]
- 54. New Freeware Offering Makes ACD/Labs Lipophilicity ... [https://www.technologynetworks.com/drug-discovery/product-news/new-freeware-offering-makes-acdlabs-lipophilicity-predictions-available-to-all-220675]
- 55. Application of Method Suitability for Drug Permeability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2976984/]
- 56. Prodrug Approach as a Strategy to Enhance Drug Permeability [https://pmc.ncbi.nlm.nih.gov/articles/PMC11946379/]
- 57. Safety and efficacy of substance-based medical devices [https://www.frontiersin.org/journals/drug-safety-and-regulation/articles/10.3389/fdsfr.2023.1124873/full]
- 58. Permeability Benchmarking: Guidelines for Comparing in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11815851/]
- 59. Advancements in techniques for analyzing cell permeability [https://www.sciencedirect.com/science/article/pii/S2405844025022236]
- 60. Recent Advances in Studying In Vitro Drug Permeation ... [https://www.mdpi.com/1999-4923/17/2/256]
- 61. Permeation Testing [https://www.teledynelabs.com/products/diffusion/permeation-testing]
- 62. LogD7.4 prediction enhanced by transferring knowledge from ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00754-4]
- 63. A systematic study of key elements underlying molecular ... [https://www.nature.com/articles/s41467-023-41948-6]
- 64. Lipophilicity prediction of peptides and peptide derivatives by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6151477/]
- 65. Enhancing molecular property prediction with quantized GNN ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-00989-3]
- 66. Scientific AI in Drug Discovery [https://www.eyesopen.com/scientific-ai-in-drug-discovery]
- 67. Molecular property prediction in the ultra‐low data regime [https://www.nature.com/articles/s42004-025-01592-1]
- 68. How to Evaluate Lipophilicity Rapidly? The Significance of ... [https://dmpkservice.wuxiapptec.com/blogs/91-how-to-evaluate-lipophilicity-rapidly-the-significance-of-reversed-phase-liquid-chromatography-rp-hplc/]
- 69. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 70. Obtaining Vialed and Plated Compounds from the NCI open ... [https://dctd.cancer.gov/drug-discovery-development/reagents-materials/vialed-plated-compounds-0]
- 71. Meta-Analysis of Permeability Literature Data Shows ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11881145/]
- 72. Multi-parameter optimization: The delicate balancing act of ... [https://www.drugdiscoverynews.com/multi-parameter-optimization-the-delicate-balancing-act-of-drug-discovery-5401]
- 73. Combining Ligand- and Structure-Based Methods for More ... [https://www.genengnews.com/topics/drug-discovery/combining-ligand-and-structure-based-methods-for-more-effective-virtual-screening/]
- 74. In Search of Beautiful Molecules: A Perspective on Generative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12458704/]