3D-QSAR in Drug Discovery: A Comprehensive Guide to CoMFA and CoMSIA Protocols
This article provides a comprehensive guide to three-dimensional quantitative structure-activity relationship (3D-QSAR) methodologies, focusing on the foundational principles, protocols, and applications of Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular...
3D-QSAR in Drug Discovery: A Comprehensive Guide to CoMFA and CoMSIA Protocols
Abstract
This article provides a comprehensive guide to three-dimensional quantitative structure-activity relationship (3D-QSAR) methodologies, focusing on the foundational principles, protocols, and applications of Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA). Tailored for researchers, scientists, and drug development professionals, it covers the entire workflow from data collection and molecular alignment to model building, validation, and troubleshooting. By integrating foundational knowledge with advanced methodological applications, practical optimization strategies, and robust validation techniques, this resource serves as a practical handbook for leveraging these powerful computational tools to accelerate rational drug design and lead optimization in biomedical research.
Understanding 3D-QSAR: From 2D Descriptors to Molecular Interaction Fields
Quantitative Structure-Activity Relationship (QSAR) methodologies represent cornerstone approaches in rational drug design. While traditional 2D-QSAR describes molecular properties using scalar parameters such as logP, molar refractivity, or electronic parameters [1], 3D-QSAR advances this paradigm by establishing relationships between biological activity and three-dimensional structural features of molecules [2]. This evolution is critically important because molecular binding occurs in three-dimensional space, with biological receptors perceiving ligands not as sets of atoms and bonds, but as specific shapes carrying complex force fields [2].
The fundamental limitation of 2D-QSAR lies in its inability to account for the spatial orientation of molecular features essential for binding interactions. 3D-QSAR addresses this by analyzing Molecular Interaction Fields (MIFs) surrounding compounds, providing a more comprehensive framework for understanding structure-activity relationships [2]. These fields quantify the steric, electrostatic, and hydrophobic interactions that govern ligand-receptor recognition, offering insights that extend beyond what classical 2D descriptors can provide [3] [2].
Fundamental Principles of 3D-QSAR
Core Theoretical Concepts
3D-QSAR operates on the principle that the biological activity of a ligand depends on its complementary interaction with a receptor binding site, mediated through various non-covalent forces [2]. The methodology systematically correlates these interaction potentials with measured biological responses through statistical models.
The approach typically involves several key steps:
- Molecular Alignment: Proper superposition of molecular structures based on their presumed pharmacophoric elements
- Interaction Field Calculation: Computation of steric, electrostatic, and other relevant potentials at grid points surrounding the molecules
- Statistical Correlation: Application of multivariate statistical methods to derive relationships between interaction fields and biological activity
A critical conceptual framework in 3D-QSAR is the probe concept, where specific molecular interaction fields are measured using representative probe atoms or groups placed at grid points throughout the molecular space [2]. Common probes include sp³ carbon atoms with +1 charge for electrostatic fields and neutral carbon atoms for steric fields [4] [2].
Molecular Interaction Fields (MIFs) and Their Significance
Molecular Interaction Fields form the descriptive foundation of 3D-QSAR models. These fields quantitatively represent how a molecule would interact with a receptor through different physicochemical forces [2]. The primary MIFs include:
- Steric Fields: Characterize repulsive and attractive van der Waals interactions, typically calculated using Lennard-Jones 6-12 potential functions [4] [2]
- Electrostatic Fields: Describe Coulombic interactions between charged or polar groups, calculated using Coulomb's law [2]
- Hydrophobic Fields: Represent the propensity for hydrophobic interactions, derived from atom-based parameters [1]
- Hydrogen Bond Donor/Acceptor Fields: Quantify the capacity for hydrogen bond formation [1]
These fields are calculated at thousands of grid points surrounding the aligned molecules, generating extensive datasets that require specialized statistical treatment through methods like Partial Least Squares (PLS) regression [3].
Comparative Analysis: 3D-QSAR vs. 2D-QSAR
Table 1: Fundamental differences between 2D-QSAR and 3D-QSAR approaches
| Feature | 2D-QSAR | 3D-QSAR |
|---|---|---|
| Molecular Representation | Scalar physicochemical parameters | 3D interaction fields in spatial grid |
| Descriptors | logP, MR, Es, σ, π, etc. [1] | Steric, electrostatic, hydrophobic potentials at grid points [2] |
| Spatial Awareness | No explicit 3D structural consideration | Explicit 3D molecular alignment required |
| Information Density | Limited number of descriptors | Thousands of field values per molecule |
| Interpretation | Mathematical coefficients in equations | 3D contour maps visualizing favorable/unfavorable regions |
| Structural Guidance | General trends for substituents | Specific spatial regions for modification |
| Receptor Insight | Indirect, implied | Indirect binding site characteristics |
The transition from 2D to 3D-QSAR represents a paradigm shift from correlative statistics to spatially informative modeling. While 2D-QSAR employs mathematical relationships like Activity = A×P1 + B×P2 + C (where P1 and P2 are physicochemical properties) [1], 3D-QSAR utilizes complex spatial datasets that provide visual guidance for molecular optimization [2]. This dimensional expansion comes with increased computational demands but offers significantly enhanced mechanistic insights into ligand-receptor interactions.
Key Methodological Approaches in 3D-QSAR
Established 3D-QSAR Techniques
Several computational methodologies have been developed to implement the 3D-QSAR paradigm, each with distinctive approaches to capturing and analyzing molecular interaction fields:
CoMFA (Comparative Molecular Field Analysis): The pioneering 3D-QSAR method that calculates steric and electrostatic interaction energies using a probe atom at grid points surrounding aligned molecules [3]. It employs Lennard-Jones and Coulomb potentials and correlates these fields with biological activity using PLS regression [3].
CoMSIA (Comparative Molecular Similarity Indices Analysis): An extension of CoMFA that calculates similarity indices using Gaussian-type distance functions, avoiding singularities at atomic positions [3]. CoMSIA typically evaluates five fields: steric, electrostatic, hydrophobic, and hydrogen bond donor and acceptor [1] [3].
GRID: A structure-based approach developed by Peter Goodford that uses diverse chemical probes to identify energetically favorable interaction sites on molecules of known structure [2]. GRID employs a smoother 6-4 potential function compared to CoMFA's Lennard-Jones potential [3].
Other Methods: Additional approaches include Molecular Shape Analysis (MSA) [3], HASL (Hypothetical Active Site Lattice) [3] [2], and GRIND (GRID INdependent Descriptors) [3], each offering unique advantages for specific applications.
Machine Learning-Enhanced 3D-QSAR
Recent advances integrate traditional 3D-QSAR with machine learning algorithms, significantly improving predictive performance. Studies demonstrate that ML-based 3D-QSAR models using Random Forest (RF), Support Vector Machine (SVM), and Multilayer Perceptron (MLP) can outperform conventional approaches in accuracy, sensitivity, and selectivity [5]. Modern implementations leverage 3D molecular similarity through shape and electrostatic comparison tools like ROCS and EON as feature inputs to ML models [6].
Experimental Protocols and Validation Standards
Protocol for CoMFA/CoMSIA Model Development
Table 2: Key validation parameters for 3D-QSAR models
| Validation Parameter | Threshold | Interpretation |
|---|---|---|
| q² (LOO cross-validation) | > 0.5 | Internal predictive ability [4] [7] |
| R² | > 0.6 | Goodness of fit for training set [4] |
| R²pred | > 0.5 | External predictive ability for test set |
| ONC (Optimal Number of Components) | - | Prevents model overfitting [4] |
| F-value | Higher preferred | Statistical significance of model |
| rm² | > 0.5 | Additional validation metric [4] |
| k, k' | 0.85-1.15 | Slope of regression line [4] |
A robust protocol for developing 3D-QSAR models involves sequential steps that ensure statistical reliability and predictive utility:
Dataset Curation and Preparation: Compile structurally diverse compounds with consistent biological activity data (e.g., IC₅₀, Ki). Convert concentration values to pIC₅₀ or pKi values for modeling [1] [8]. Divide compounds into training (typically 80-85%) and test sets (15-20%) [9] [7].
Molecular Modeling and Alignment: Generate energetically optimized 3D structures using tools like LigPrep [9] with appropriate force fields (e.g., OPLS_2005) [9]. Perform molecular alignment based on common pharmacophoric features or scaffold superimposition [7].
Interaction Field Calculation: For CoMFA, calculate steric (Lennard-Jones 6-12 potential) and electrostatic (Coulomb potential) fields using an sp³ carbon probe with +1 charge at grid points with 2.0 Å spacing [4]. For CoMSIA, compute similarity indices for steric, electrostatic, hydrophobic, and hydrogen bond donor/acceptor fields using a Gaussian-type function [3].
Statistical Analysis and Validation: Apply Partial Least Squares (PLS) regression to correlate interaction fields with biological activity [4] [7]. Perform leave-one-out (LOO) cross-validation to determine the optimal number of components and q² value [4]. Validate models using external test sets, bootstrapping, and progressive scrambling techniques [4] [8].
Diagram 1: 3D-QSAR modeling workflow showing the sequential protocol from dataset preparation to application in compound design.
Table 3: Essential computational tools and resources for 3D-QSAR studies
| Tool/Resource | Type | Primary Function |
|---|---|---|
| Schrödinger Suite | Commercial Software | Comprehensive drug discovery platform with LigPrep [9], Phase [9] |
| SYBYL | Commercial Software | Original CoMFA/CoMSIA implementation [3] |
| GRID | Commercial Software | Molecular interaction field calculations [3] [2] |
| OpenEye Orion | Commercial Platform | 3D-QSAR with machine learning integration [6] |
| VMD with APBS Plugin | Open Source | Molecular visualization and electrostatic potential calculation [2] |
| PLS Regression | Statistical Method | Multivariate correlation of fields with activity [3] |
| LOO Cross-Validation | Validation Method | Internal model validation [4] |
Applications and Case Studies in Drug Discovery
Successful Implementations Across Target Classes
3D-QSAR methodologies have demonstrated significant utility across diverse therapeutic targets:
Kinase Inhibitors: CoMFA and CoMSIA models were developed for pyrimidine-based JAK3 inhibitors, resulting in highly predictive models (q² = 0.717, r² = 0.986) that guided the design of novel compounds with improved potency [8]. Similarly, 3D-QSAR informed the design of Bcr-Abl inhibitors to overcome resistance mutations in chronic myeloid leukemia treatment [10].
Epigenetic Targets: For mutant isocitrate dehydrogenase 1 (mIDH1) inhibitors, 3D-QSAR models (CoMFA: q² = 0.765, R² = 0.980; CoMSIA: q² = 0.770, R² = 0.997) enabled rational design of novel pyridin-2-one derivatives with predicted enhanced activity [7].
Tubulin-Targeting Agents: Pharmacophore-based 3D-QSAR on cytotoxic quinolines identified a six-point hypothesis (AAARRR.1061) with three hydrogen bond acceptors and three aromatic rings, demonstrating high correlation (R² = 0.865) and guiding virtual screening efforts [9].
Endocrine Disruptor Screening: Machine learning-based 3D-QSAR models were developed to predict estrogen receptor-binding activity of small molecules, outperforming traditional VEGA models in accuracy, sensitivity, and selectivity for endocrine disruption assessment [5].
Integration with Complementary Computational Approaches
Modern 3D-QSAR is frequently integrated with other structure-based methods in synergistic workflows:
3D-QSAR with Molecular Docking: Combined approaches leverage docking-generated alignments for 3D-QSAR while using 3D-QSAR results to optimize docking scores through focused library design [3] [8].
3D-QSAR with Molecular Dynamics: MD simulations validate 3D-QSAR predictions by assessing binding stability and calculating binding free energies through MM/PBSA approaches [8] [7].
3D-QSAR with ADMET Prediction: Integration of activity predictions with absorption, distribution, metabolism, excretion, and toxicity profiling enables comprehensive compound optimization [8] [7].
Diagram 2: 3D-QSAR integration with complementary computational and experimental methods in drug discovery workflows.
The evolution of 3D-QSAR continues through integration with emerging computational technologies. Machine learning enhancement represents the most significant advancement, with algorithms capable of learning complex patterns from 3D molecular features to improve predictive accuracy [5] [6]. Modern implementations provide prediction confidence estimates, guiding researchers on when to trust model outputs and when to employ more rigorous physics-based methods like free energy calculations [6].
Further development is expected in several key areas:
- Automated model interpretation that directly suggests structural modifications
- Enhanced treatment of molecular flexibility through 4D and 5D QSAR approaches
- Integration with structural biology data from cryo-EM and molecular dynamics
- Application to novel modalities beyond small molecules, including peptides and fragment-based designs
In conclusion, 3D-QSAR has evolved substantially beyond traditional 2D approaches by explicitly incorporating the spatial dimensions central to molecular recognition. Through continuous methodological refinements and integration with complementary computational techniques, 3D-QSAR maintains its critical role in modern rational drug design, enabling researchers to efficiently navigate chemical space and optimize compound properties with structural insight.
Core Principles of CoMFA (Comparative Molecular Field Analysis)
Comparative Molecular Field Analysis (CoMFA) is a cornerstone methodology in modern computational drug discovery, representing a significant advancement in three-dimensional quantitative structure-activity relationship (3D-QSAR) modeling. Unlike traditional 2D-QSAR approaches that utilize numerical molecular descriptors, CoMFA characterizes molecules based on their three-dimensional interaction fields with probe atoms, providing a more comprehensive representation of molecular properties critical to biological activity [11]. This technique has become an indispensable tool for researchers and medicinal chemists seeking to understand the intricate relationship between molecular structure and biological effect, ultimately guiding the rational design of novel therapeutic compounds with enhanced potency and selectivity [12].
The fundamental premise of CoMFA rests on the concept that a molecule's biological activity is determined by its steric (shape-related) and electrostatic (charge-related) properties in three-dimensional space. By quantitatively analyzing how these molecular fields correlate with measured biological responses across a series of compounds, CoMFA generates predictive models and intuitive visual maps that pinpoint specific chemical features responsible for activity variations [11]. These insights are particularly valuable in optimizing lead compounds, as they directly suggest where and what type of structural modifications may enhance desired biological interactions.
Fundamental Principles and Theoretical Basis
The theoretical foundation of CoMFA is built upon several key principles that differentiate it from conventional QSAR approaches. First, it operates on the bioactive conformation assumption, positing that molecules must be analyzed in their three-dimensional orientations that correspond to how they bind to biological targets [11]. Second, it employs the molecular field analogy, which suggests that non-covalent interaction forces between a ligand and its receptor can be sampled using probe atoms placed around the molecular surface. Finally, it utilizes statistical correlation methods to establish quantitative relationships between these sampled field values and biological activity measurements.
A critical advancement over traditional methods is CoMFA's ability to handle the high-dimensional descriptor space inherent in 3D molecular representations. While classical QSAR uses a compact set of global molecular descriptors that are invariant to molecular conformation and orientation, CoMFA descriptors are derived directly from the spatial structure of the molecule and are therefore sensitive to its three-dimensional arrangement [11]. This provides a much finer resolution of molecular interactions but introduces challenges related to molecular alignment and data dimensionality that must be carefully addressed during model development.
Experimental Protocol and Workflow
The standard CoMFA methodology follows a systematic, multi-stage workflow that transforms raw molecular structures into validated predictive models. Each stage requires careful execution to ensure the resulting model is both statistically robust and chemically meaningful.
Data Collection and Preparation
The initial stage involves assembling a homogeneous dataset of compounds with experimentally determined biological activities (typically IC₅₀, EC₅₀, or Kᵢ values) measured under consistent conditions [11]. The integrity of this dataset is paramount, as variability in assay protocols introduces noise and systemic bias that compromise predictive value. The dataset should contain sufficient structural diversity to capture meaningful structure-activity relationships while maintaining enough similarity to assume a common binding mode. Typically, 20-50 compounds are required, with 25-33% reserved as an external test set for validation [12].
Molecular Modeling and Alignment
With the dataset defined, 2D molecular structures are converted to 3D coordinates using cheminformatics tools like RDKit or Sybyl, then geometry-optimized using molecular mechanics force fields (e.g., Tripos Force Field) or quantum mechanical methods to ensure realistic, low-energy conformations [11] [12]. The most critical step—molecular alignment—involves superimposing all molecules within a shared 3D reference frame that reflects their putative bioactive conformations [11]. This can be achieved through:
- Common substructure alignment: Based on shared scaffolds or maximum common substructures
- Pharmacophore alignment: Using tools like GALAHAD to align molecules according to hypothesized pharmacophoric features [12]
- Docking-based alignment: Using predicted binding orientations from molecular docking
Table 1: Molecular Alignment Methods in CoMFA
| Method | Description | Applications |
|---|---|---|
| Bemis-Murcko Scaffold | Defines core structure by removing side chains, retaining ring systems and linkers | Widely used for clustering and scaffold-based analysis of congeneric series [11] |
| Maximum Common Substructure (MCS) | Identifies largest substructure shared among molecules | Useful for comparing diverse chemotypes when clear scaffolds are not defined [11] |
| Pharmacophore-Based | Aligns molecules based on common pharmacophoric features | Superior for datasets with limited structural commonality [12] |
Field Calculation and Descriptor Generation
Following alignment, molecules are placed within a 3D cubic lattice with typical grid spacing of 1.0-2.0 Å in each dimension [12]. A probe atom (typically an sp³ carbon with +1 charge) is placed at each grid point to calculate steric (Lennard-Jones) and electrostatic (Coulombic) interaction energies with the molecule [12]. An energy cutoff value (typically 30 kcal/mol) is applied to avoid unrealistic energy values near molecular surfaces [12]. This process generates thousands of field values for each compound, creating the high-dimensional descriptor matrix for subsequent statistical analysis.
Statistical Analysis and Model Validation
The relationship between CoMFA field descriptors and biological activity is established using Partial Least Squares (PLS) regression, which handles the large number of correlated descriptors by projecting them to a smaller set of latent variables [11] [12]. Model validation employs:
- Leave-One-Out (LOO) cross-validation: To determine the optimal number of components and calculate q² (cross-validated correlation coefficient) [12]
- External validation: Using the reserved test set to calculate r²pred (predictive correlation coefficient) [12]
- Goodness-of-fit: Assessed by conventional r² (non-cross-validated correlation coefficient)
A robust CoMFA model typically exhibits q² > 0.5 and r²pred > 0.6, indicating both internal consistency and predictive capability for new compounds [12].
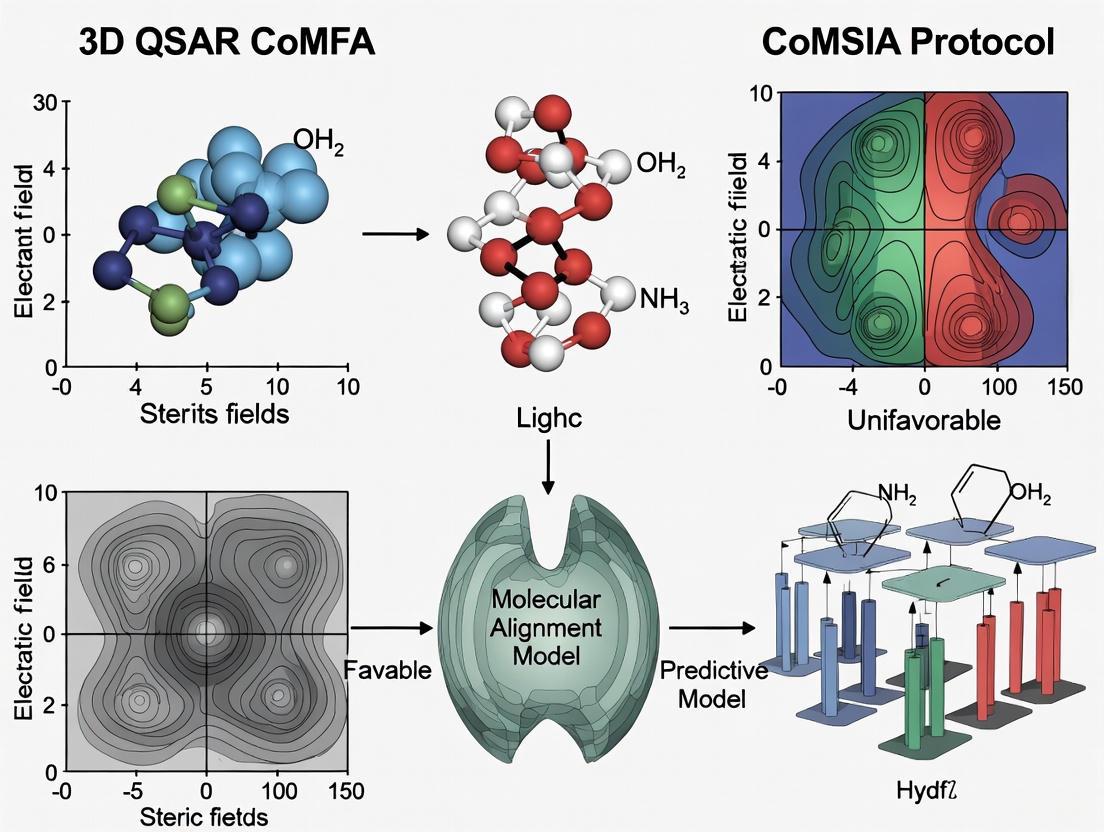
Model Interpretation and Visualization
The final CoMFA model is interpreted through contour maps that identify spatial regions where specific molecular features enhance or diminish biological activity [11]. These maps are visualized overlaying a reference compound:
- Green contours: Indicate regions where increased steric bulk enhances activity
- Yellow contours: Indicate regions where decreased steric bulk enhances activity
- Blue contours: Indicate regions where positive electrostatic potential enhances activity
- Red contours: Indicate regions where negative electrostatic potential enhances activity
These visualizations translate complex statistical models into intuitive chemical guidance, directly suggesting structural modifications to optimize activity [11].
Workflow Visualization
The following diagram illustrates the comprehensive CoMFA workflow, from initial data preparation through to model application in drug design:
Comparative Analysis: CoMFA vs. CoMSIA
Comparative Molecular Similarity Indices Analysis (CoMSIA) represents an extension and refinement of the CoMFA methodology. While both approaches share similar conceptual foundations, they differ significantly in their technical implementation and practical applications.
Table 2: Comparison of CoMFA and CoMSIA Approaches
| Feature | CoMFA | CoMSIA |
|---|---|---|
| Field Calculation | Uses Lennard-Jones (steric) and Coulombic (electrostatic) potentials with a probe atom on a 3D grid [11] [12] | Uses Gaussian-type similarity functions to compute multiple molecular fields [11] |
| Fields Included | Primarily steric and electrostatic fields [12] | Steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields [11] [12] |
| Alignment Sensitivity | Highly sensitive to molecular alignment; precise alignment is crucial for reliable models [11] | More robust to small changes in alignment, suitable for structurally diverse datasets [11] |
| Distance Dependence | Potential energy values show abrupt changes near molecular surfaces [11] | Smoother distance dependence due to Gaussian functions; no arbitrary cutoff needed [11] |
| Applications | Best for congeneric series with reliable alignment; provides clear steric/electrostatic interpretation [12] | Superior for structurally diverse datasets; offers additional hydrophobic and H-bonding insights [11] |
Advanced Applications and Recent Developments
The application of CoMFA has expanded significantly since its introduction, with recent advancements incorporating machine learning algorithms to enhance predictive performance. Studies have demonstrated that 3D-QSAR models utilizing random forest (RF), support vector machine (SVM), and multilayer perceptron (MLP) algorithms can outperform traditional statistical methods in terms of accuracy, sensitivity, and selectivity [5]. These hybrid approaches leverage the rich descriptor space of CoMFA while benefiting from the pattern recognition capabilities of machine learning.
Recent research applications highlight CoMFA's continued relevance in addressing contemporary drug discovery challenges:
- Bcr-Abl inhibitors for leukemia treatment: 3D-QSAR models successfully guided the design of purine derivatives with enhanced potency against both wild-type and mutant Bcr-Abl, including the recalcitrant T315I mutation [10]
- α1A-Adrenergic receptor antagonists: CoMFA and CoMSIA studies based on pharmacophore molecular alignment yielded robust models (q² = 0.840) that identified key electrostatic, hydrophobic, and hydrogen bonding interactions governing antagonist activity [12]
- Estrogen receptor-binding activity prediction: Machine learning-based 3D-QSAR models demonstrated superior accuracy and sensitivity compared to conventional VEGA models for predicting endocrine disruption potential of new chemical entities [5]
These applications underscore CoMFA's versatility across diverse target classes and its adaptability through integration with complementary computational approaches.
Successful implementation of CoMFA studies requires access to specialized software tools, computational resources, and methodological expertise. The following table outlines key components of the CoMFA research toolkit:
Table 3: Essential Resources for CoMFA Research
| Resource Category | Specific Tools/Resources | Function/Purpose |
|---|---|---|
| Molecular Modeling Software | SYBYL/Tripos, RDKit, Open3DALIGN | Generation of 3D structures, energy minimization, conformational analysis, and molecular alignment [11] [12] |
| CoMFA/CoMSIA Platforms | SYBYL CoMFA Module, Open3DQSAR | Calculation of steric/electrostatic fields, PLS regression, contour map generation [12] |
| Alignment Tools | GALAHAD, Phase, ROCS | Pharmacophore-based alignment, maximum common substructure identification [12] |
| Statistical Analysis | R, Python (scikit-learn), MATLAB | Partial least squares regression, cross-validation, model validation [11] |
| Visualization Software | PyMOL, Chimera, VMD | Visualization of molecular structures, contour maps, and binding interactions [11] |
Best Practices and Methodological Considerations
To ensure the development of robust and predictive CoMFA models, researchers should adhere to several established best practices:
- Data Quality Assurance: Verify consistency of biological activity measurements and chemical structure curation before analysis [11]
- Comprehensive Validation: Employ both internal (cross-validation) and external (test set prediction) validation methods [12]
- Statistical Significance: Report relevant statistical metrics including q², r², standard error of estimate, and F-value [12]
- Chemical Interpretation: Relate contour maps to known structural and mechanistic information to ensure chemical plausibility [11]
- Applicability Domain: Clearly define the structural space where the model provides reliable predictions
Proper implementation of these practices mitigates common pitfalls such as overfitting, chance correlations, and inaccurate extrapolation beyond the model's training domain.
Comparative Molecular Field Analysis remains a fundamentally important approach in modern drug discovery, providing a powerful framework for understanding three-dimensional structure-activity relationships. Its unique ability to transform complex molecular interaction data into visually interpretable contour maps makes it particularly valuable for medicinal chemists seeking to optimize lead compounds. When properly implemented with careful attention to alignment, validation, and interpretation, CoMFA and its CoMSIA extension continue to deliver impactful insights that accelerate the development of novel therapeutic agents across diverse disease areas.
The ongoing integration of CoMFA with emerging machine learning methodologies promises to further enhance its predictive power and application scope, ensuring its continued relevance in an increasingly data-driven drug discovery landscape. As computational resources expand and algorithmic sophistication increases, CoMFA-based approaches will likely play an increasingly central role in bridging the gap between molecular structure and biological function.
Core Principles of CoMSIA (Comparative Molecular Similarity Indices Analysis)
Comparative Molecular Similarity Indices Analysis (CoMSIA) is a sophisticated ligand-based, alignment-dependent 3D-QSAR method that serves as a modified and advanced version of Comparative Molecular Field Analysis (CoMFA) [13]. This technique was introduced to address several limitations inherent in the CoMFA approach, primarily its high sensitivity to molecular alignment and the abrupt changes in grid-based probe-atom interactions [14]. CoMSIA achieves this by employing Gaussian-type distance-dependent functions instead of the traditional Lennard-Jones and Coulomb potentials used in CoMFA, resulting in smoother sampling of the molecular fields and more interpretable contour maps [13] [14].
The fundamental concept of CoMSIA revolves around analyzing molecular similarity indices calculated using a probe atom at regularly spaced grid intersections surrounding an aligned set of molecules [14]. Unlike CoMFA, which primarily focuses on steric and electrostatic fields, CoMSIA extends the analysis to include hydrophobic and hydrogen-bonding properties, providing a more comprehensive description of the interactions responsible for ligand binding [13] [15]. This multi-field approach allows researchers to capture a broader spectrum of the physicochemical properties that influence biological activity, including solvent entropic effects through the hydrophobic probe [14].
Core Principles and Theoretical Foundation
Molecular Similarity Fields
CoMSIA calculates similarity indices using a common probe atom with specific properties that is placed at regularly spaced grid points surrounding the aligned molecules [13]. The method typically employs five distinct molecular fields to characterize the physicochemical properties of the molecules under investigation [13] [14]:
- Steric fields (representing molecular bulk and shape)
- Electrostatic fields (representing charge distribution)
- Hydrophobic fields (representing lipophilicity)
- Hydrogen bond donor fields
- Hydrogen bond acceptor fields
The similarity indices (AF,k) for each molecule j with atoms i at grid point q are calculated using a Gaussian-type function [16]:
A_F,k(q) = -Σ[wprobe,k × wik × e^(-α×r²iq)]
where wprobe,k represents the probe value for property k, wik is the actual value of the property for atom i, riq is the distance between the probe and atom i, and α is the attenuation factor [16].
Advantages Over CoMFA
CoMSIA offers several distinct advantages that address key limitations of the CoMFA approach [13] [14]:
- Avoidance of Singularities: The Gaussian function eliminates the abrupt changes in potential energy that occur near molecular surfaces in CoMFA, providing smoother and more continuous fields [14].
- Broader Property Coverage: The inclusion of hydrophobic and explicit hydrogen-bonding fields enables a more comprehensive characterization of ligand-receptor interactions [15].
- Enhanced Interpretability: The resulting contour maps more intuitively indicate regions where specific physicochemical properties enhance or diminish biological activity [14] [15].
- Solvent Effect Modeling: The hydrophobic field attempts to capture solvent entropic terms, providing a more realistic representation of the binding environment [14].
Table 1: Key Differences Between CoMFA and CoMSIA Approaches
| Feature | CoMFA | CoMSIA |
|---|---|---|
| Potential Functions | Lennard-Jones and Coulomb potentials [13] | Gaussian-type similarity functions [13] |
| Fields Calculated | Steric and electrostatic [11] | Steric, electrostatic, hydrophobic, H-bond donor, H-bond acceptor [13] |
| Alignment Sensitivity | Highly sensitive [11] | More robust to minor misalignments [11] |
| Contour Maps | Highlight regions where molecules interact with receptor environment [13] | Indicate areas within ligand region that favor/dislike specific properties [13] |
| Probe Atom Properties | sp³ carbon with +1 charge for steric/electrostatic fields [16] | Radius 1Å, charge +1, hydrophobicity +1, H-bond donor/acceptor +1 [16] |
CoMSIA Methodology and Protocols
Molecular Preparation and Alignment
The initial steps in CoMSIA involve careful preparation and alignment of the molecular dataset [11]:
- Structure Generation: Convert 2D molecular structures to 3D coordinates using molecular modeling software such as SYBYL [16].
- Geometry Optimization: Energy minimization of the molecules using force fields (e.g., Tripos force field) or quantum mechanical methods [16]. The Powell conjugate gradient algorithm with a convergence criterion of 0.001 kcal/(mol·Å) is typically employed [16].
- Partial Charge Calculation: Compute atomic partial charges using methods such as Gasteiger-Hückel [16].
- Molecular Alignment: Superimpose molecules based on a common template or pharmacophore hypothesis. The most active molecule is often used as a template, and alignment can be achieved through field-fit methods or maximum common substructure (MCS) approaches [13] [11].
Field Calculation and Model Development
Following molecular alignment, the CoMSIA fields are calculated and analyzed [13]:
- Grid Generation: A 3D grid box is created around the aligned molecules, typically extending 2.0 Å beyond the molecular dimensions in all directions [13].
- Similarity Index Calculation: The five CoMSIA fields (steric, electrostatic, hydrophobic, H-bond donor, H-bond acceptor) are computed at each grid point using a common probe atom with specific properties [13].
- Partial Least Squares (PLS) Analysis: The relationship between the similarity indices (descriptors) and biological activity is established using PLS regression [13]. The PLS algorithm projects the numerous correlated descriptors into a smaller set of latent variables that best explain the variance in biological activity [11].
- Model Validation: The robustness and predictive ability of the model are assessed using cross-validation techniques (e.g., leave-one-out) and an external test set [17]. Statistical metrics such as R² (goodness-of-fit), Q² (cross-validated correlation coefficient), and R²pred (predictive ability for test set) are calculated [17].
Contour Map Generation and Interpretation
The final step involves generating and interpreting contour maps that visualize the relationship between molecular properties and biological activity [14]:
- Contour Map Calculation: Coefficient values from the PLS analysis are contoured to identify regions where specific molecular properties correlate with enhanced or diminished biological activity [14].
- Map Interpretation: The contour maps are superimposed on the molecular structures to guide rational drug design [11]:
- Green steric contours indicate regions where bulky groups enhance activity.
- Yellow steric contours indicate regions where bulky groups diminish activity.
- Blue electrostatic contours indicate regions where positive charges enhance activity.
- Red electrostatic contours indicate regions where negative charges enhance activity.
Research Reagent Solutions and Essential Materials
Table 2: Essential Research Reagents and Computational Tools for CoMSIA Studies
| Tool/Reagent | Function/Application | Typical Specifications |
|---|---|---|
| SYBYL Molecular Modeling Software | Primary platform for CoMFA/CoMSIA studies [16] | Includes modules for structure building, minimization, alignment, and field calculation [16] |
| Tripos Force Field | Energy minimization of molecular structures [16] | Distance-dependent dielectric, Powell conjugate gradient algorithm [16] |
| Gasteiger-Hückel Method | Calculation of partial atomic charges [16] | Rapid approximate calculation of charge distribution [16] |
| PLS Algorithm | Statistical correlation of fields with biological activity [13] | Handles multiple correlated descriptors through latent variables [11] |
| Probe Atoms | Calculation of similarity indices at grid points [13] | Radius: 1Å, Charge: +1, Hydrophobicity: +1, H-bond properties: +1 [13] |
Applications and Case Studies
CoMSIA has been successfully applied to various drug discovery programs, demonstrating its utility in rational drug design. In one notable application, CoMSIA was used to study thermolysin inhibitors, where the method provided significantly improved and easily interpretable contour maps compared to CoMFA [15]. The features highlighted in the CoMSIA maps intuitively suggested where to modify molecular structures in terms of physicochemical properties and functional groups to improve binding affinity [15]. Furthermore, the derived correlation model was used to score different members of a combinatorial library designed for thermolysin inhibition, demonstrating the predictive power of the CoMSIA method [15].
In another study on phenyl alkyl ketones as phosphodiesterase 4 inhibitors, CoMSIA models demonstrated high predictive ability with R²(pred) values of 0.9470 [17]. The models were developed based on pharmacophore alignment and exhibited robust statistical characteristics, enabling the design of novel molecules with predicted high activity that also passed Lipinski's rule of five for drug-likeness [17].
Table 3: Statistical Performance Metrics from Representative CoMSIA Studies
| Study/Application | Q² (Cross-validated) | R² (Conventional) | R²pred (Predictive) | Fields Used |
|---|---|---|---|---|
| Phenyl alkyl ketones as PDE4 inhibitors [17] | 0.8539 | 0.9610 | 0.9470 | Steric, Electrostatic, Hydrophobic, H-bond Donor, H-bond Acceptor |
| Thermolysin inhibitors (Reference study) [15] | Comparable to CoMFA | Comparable to CoMFA | High prediction power | Steric, Electrostatic, Hydrophobic, H-bond Donor, H-bond Acceptor |
Advanced Protocols and Methodological Considerations
Region Focusing and Variable Selection
To enhance the quality of CoMSIA models, several advanced techniques can be employed:
- Region Focusing: This technique refines the model by improving the weight for lattice points that are most relevant to the model, thereby enhancing the contribution of these significant points [16].
- Statistical Validation: The robustness of CoMSIA models should be confirmed using leave-one-out cross-validation, while the predictive ability should be tested using an external test set [17].
- Field Combination Strategies: Testing all possible combinations of different fields to acquire optimal predictive CoMSIA models is essential [16]. The standard CoMSIA settings include a column filtering of 0.3 kcal/mol (instead of the default 2 kcal/mol) to reduce noise and attenuate the signal-to-noise ratio [16].
Integration with Other Computational Methods
CoMSIA is often used in conjunction with other computational approaches to enhance its predictive power and applicability:
- Docking Studies: CoMSIA results can be integrated with molecular docking to validate the binding mode of designed molecules and correlate predicted activity with docking scores [17].
- Pharmacophore Modeling: CoMSIA models can be developed based on alignment obtained from 3D pharmacophore models, providing a more biologically relevant superposition of molecules [17].
- ADMET Filtering: Designed molecules can be evaluated using Lipinski's rule of five and other drug-likeness filters to ensure pharmaceutical relevance [17].
The CoMSIA methodology represents a significant advancement in 3D-QSAR techniques, offering improved interpretability and a more comprehensive characterization of molecular interactions essential for rational drug design. Its ability to incorporate multiple physicochemical properties and generate intuitively understandable contour maps makes it an invaluable tool in modern medicinal chemistry and drug discovery programs.
Key Differences Between CoMFA and CoMSIA Methodologies
In the field of computer-aided drug design, three-dimensional quantitative structure-activity relationship (3D-QSAR) methods are pivotal for understanding how the structural and physicochemical properties of molecules correlate with their biological activity. Among these techniques, Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) stand out as the most widely used approaches [18]. Both methods aim to correlate 3D molecular fields with biological responses using statistical techniques like Partial Least Squares (PLS) regression. However, they differ fundamentally in how they calculate and interpret these molecular fields, leading to distinct advantages and applications. This article provides a detailed comparison of CoMFA and CoMSIA methodologies, framed within protocols for 3D-QSAR research, to guide researchers in selecting and implementing the appropriate technique for their drug discovery projects.
Conceptual Foundations and Methodological Differences
Core Principles of CoMFA
Comparative Molecular Field Analysis (CoMFA), introduced by Cramer et al. in 1988, is considered the pioneering 3D-QSAR method [13] [18]. Its fundamental hypothesis is that the biological properties of molecules can be correlated with their non-covalent interaction fields surrounding the molecule, primarily steric and electrostatic fields [13].
In CoMFA, a probe atom (typically an sp³ carbon with a +1 charge) is placed at regularly spaced grid points around a set of pre-aligned molecules. At each grid point, the steric (Lennard-Jones) and electrostatic (Coulombic) interaction energies between the probe and each molecule are calculated [13] [12]. These interaction energies serve as descriptors for subsequent PLS analysis to build a predictive QSAR model. A significant limitation of this approach is the need for energy cutoffs (typically 30 kcal/mol) to avoid unrealistic energy values near molecular surfaces, which can result in abrupt field changes and potential artifacts in the model [13] [19].
Core Principles of CoMSIA
Comparative Molecular Similarity Indices Analysis (CoMSIA) was developed by Klebe et al. as an advanced alternative to address several CoMFA limitations [13] [19]. Rather than calculating interaction energies, CoMSIA evaluates similarity indices between molecules at regularly spaced grid points using a common probe atom [13].
CoMSIA employs a Gaussian-type function to calculate these similarity indices, providing a "softer" potential without the abrupt changes characteristic of CoMFA fields [13] [19]. This approach eliminates the need for arbitrary energy cutoffs and results in more stable models that are less sensitive to molecular orientation and grid positioning [19]. Additionally, CoMSIA extends beyond the steric and electrostatic fields of CoMFA by incorporating hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields, providing a more comprehensive description of molecular interactions [13].
Table 1: Fundamental Differences Between CoMFA and CoMSIA Approaches
| Feature | CoMFA | CoMSIA |
|---|---|---|
| Field Calculation | Based on interaction energies (Lennard-Jones and Coulomb potentials) | Based on similarity indices using Gaussian-type function |
| Field Types | Steric and electrostatic | Steric, electrostatic, hydrophobic, hydrogen bond donor, hydrogen bond acceptor |
| Probe Atom | sp³ carbon with +1 charge | Similar probe with defined properties for multiple fields |
| Potential Function | "Hard" potentials with abrupt changes | "Softer" potentials with smooth distance dependence |
| Cutoff Values | Required (typically 30 kcal/mol) | Not required |
| Sensitivity to Alignment | Highly sensitive | Less sensitive |
| Interpretation | Highlights regions where interactions would occur | Indicates areas within ligand space that favor particular properties |
Comparative Statistical Performance
Both CoMFA and CoMSIA models are evaluated using similar statistical measures, including the leave-one-out cross-validated correlation coefficient (q²), non-cross-validated correlation coefficient (r²), and predictive r² for test set compounds (r²pred) [20] [12]. Generally, a model is considered statistically significant and predictive when q² > 0.5 and r² > 0.6 [20] [21].
Research applications demonstrate that both methods can produce highly predictive models, though their performance varies depending on the molecular system under investigation. For example, in a study on α1A-adrenergic receptor antagonists, both methods showed comparable predictive power with q² values of 0.840 [12]. Conversely, in a study on phenylsulfonyl carboxylates, CoMFA produced a superior model (q² = 0.823) compared to CoMSIA (q² = 0.713) [22].
Table 2: Representative Statistical Performance of CoMFA and CoMSIA from Various Studies
| Study System | CoMFA q² | CoMFA r² | CoMSIA q² | CoMSIA r² | Reference |
|---|---|---|---|---|---|
| α1A-Adrenergic Receptor Antagonists | 0.840 | N/R | 0.840 | N/R | [12] |
| Phenylsulfonyl Carboxylates | 0.823 | 0.958 | 0.713 | 0.933 | [22] |
| Thieno-Pyrimidine Derivatives (TNBC) | 0.818 | 0.917 | 0.801 | 0.897 | [21] |
| Ionone-based Chalcones (Prostate Cancer) | 0.527 | 0.636 | 0.550 | 0.671 | [20] |
| Aryloxypropanolamines (β3-AR) | 0.537 | 0.993 | 0.669 | 0.984 | [23] |
Experimental Protocols
Standardized Workflow for 3D-QSAR Studies
The following workflow outlines the general procedure for conducting both CoMFA and CoMSIA studies, with method-specific variations noted where applicable.
Protocol 1: Data Set Preparation and Molecular Alignment
Purpose: To curate a structurally and biologically diverse set of compounds and align them in 3D space based on their putative bioactive conformation.
Critical Steps:
Compound Selection: Select 20-50 congeneric compounds with:
- Quantified biological activity (IC₅₀, Kᵢ, EC₅₀) spanning 3-4 orders of magnitude
- Structural diversity with a common core scaffold
- Consistent mechanism of action and binding mode [13]
Data Set Division: Divide compounds into training (70-80%) and test (20-30%) sets, ensuring:
- Structural diversity and activity range representation in both sets
- Test set size of 25-33% of total compounds for reliable validation [12]
Molecular Modeling:
Molecular Alignment (Most Critical Step):
Protocol 2: Field Calculation and Model Development
Purpose: To calculate molecular fields and develop statistically robust 3D-QSAR models using PLS regression.
Critical Steps:
Grid Generation:
CoMFA Field Calculation:
CoMSIA Field Calculation:
- Use probe atom with charge=+1, hydrophobicity=+1, H-bond donor=+1, H-bond acceptor=+1 [13] [12]
- Calculate similarity indices using Gaussian function with attenuation factor α=0.3 [20] [19]
- Include combinations of steric (S), electrostatic (E), hydrophobic (H), hydrogen bond donor (D), and hydrogen bond acceptor (A) fields [23] [21]
PLS Analysis and Validation:
- Perform leave-one-out (LOO) cross-validation to determine optimal number of components (N) [20] [12]
- Build final model using optimal N with non-cross-validated analysis [20]
- Validate model using test set compounds and calculate predictive r² (r²pred) [20]
- Perform progressive scrambling stability test to confirm robustness [21]
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Essential Computational Tools for 3D-QSAR Studies
| Tool Category | Specific Examples | Function in 3D-QSAR |
|---|---|---|
| Molecular Modeling Software | SYBYL/Tripos, Schrödinger, MOE, OpenBabel | Core platform for molecular modeling, alignment, and field calculations |
| Open-Source Alternatives | Py-CoMSIA (Python with RDKit, NumPy) | Open-source implementation of CoMSIA methodology [19] |
| Force Fields | Tripos Force Field, MMFF94, AMBER | Energy minimization and conformational analysis |
| Charge Calculation Methods | Gasteiger-Hückel, Gasteiger, Mulliken, Del-Re | Calculation of partial atomic charges for electrostatic fields |
| Statistical Analysis | Partial Least Squares (PLS) in SYBYL, MATLAB | Correlation of field variables with biological activity |
| Visualization Tools | PyMOL, MOLCAD, PyVista | Visualization of contour maps and molecular interactions |
Applications in Drug Discovery
Case Studies and Research Applications
Both CoMFA and CoMSIA have been extensively applied across various therapeutic areas, demonstrating their utility in rational drug design:
Cancer Therapeutics: In a study on thieno-pyrimidine derivatives as triple-negative breast cancer inhibitors, CoMFA (q² = 0.818, r² = 0.917) and CoMSIA (q² = 0.801, r² = 0.897) models successfully identified key structural features for VEGFR3 inhibition [21]. The contour maps guided optimization of steric, electrostatic, and hydrophobic properties to enhance potency.
Cardiovascular Diseases: For aryloxypropanolamine compounds targeting β3-adrenergic receptors for diabetes and obesity treatment, CoMSIA models incorporating all field types showed superior predictive ability (r² = 0.918) compared to CoMFA (r² = 0.865) [23]. The hydrophobic and hydrogen bond acceptor fields provided critical insights for selectivity.
Prostate Cancer: Research on ionone-based chalcones demonstrated CoMSIA (q² = 0.550) slightly outperforming CoMFA (q² = 0.527) in predicting anti-prostate cancer activity [20]. The additional field types in CoMSIA offered more comprehensive interaction information.
Renin Inhibitors: In the design of novel renin inhibitors for cardiovascular diseases, combined CoMFA/CoMSIA studies with docking revealed key binding interactions, demonstrating the complementary nature of these approaches [24].
Contour Map Interpretation and Molecular Design
The primary output from both CoMFA and CoMSIA studies is a set of contour maps that visualize regions where specific molecular properties enhance or diminish biological activity.
CoMFA Contour Interpretation:
- Steric Fields: Green contours indicate regions where bulky groups enhance activity; yellow contours where bulky groups decrease activity [21]
- Electrostatic Fields: Blue contours indicate regions where positive charge enhances activity; red contours where negative charge enhances activity [21]
CoMSIA Contour Interpretation:
- Includes additional contours for hydrophobic fields (yellow-favorable, white-unfavorable), hydrogen bond donors (cyan-favorable, purple-unfavorable), and acceptors (magenta-favorable, red-unfavorable) [23] [21]
A key interpretive difference is that CoMFA contours highlight regions in space where the aligned molecules would favorably interact with a receptor environment, while CoMSIA contours indicate areas within the region occupied by the ligands that favor or dislike specific physicochemical properties [13]. This makes CoMSIA maps more directly useful for determining whether all features crucial for biological response are present in structures being considered for design.
CoMFA and CoMSIA represent complementary approaches in the 3D-QSAR toolkit, each with distinct advantages. CoMFA serves as the foundational method with straightforward interpretation of steric and electrostatic interaction fields. CoMSIA extends this framework with smoother potential functions, additional field types, and reduced sensitivity to alignment artifacts. The choice between methods depends on research objectives: CoMFA for straightforward steric/electrostatic analysis, CoMSIA for comprehensive interaction profiling including hydrophobic and hydrogen bonding effects. Implementation of the standardized protocols outlined herein will enable researchers to effectively apply these powerful techniques to accelerate drug discovery and optimization efforts.
Essential Software and Tools for 3D-QSAR Studies
Three-dimensional Quantitative Structure-Activity Relationship (3D-QSAR) represents a significant advancement over traditional 2D-QSAR methods by incorporating spatial molecular features to build predictive models. These techniques are crucial in modern drug discovery for elucidating the complex relationships between the three-dimensional structural properties of molecules and their biological activities. Among the most established 3D-QSAR methodologies are Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA). CoMFA operates by calculating steric (Lennard-Jones potential) and electrostatic (Coulombic potential) interaction energies between a probe atom and aligned molecules at regularly spaced grid points [16] [25]. CoMSIA extends this approach by incorporating additional similarity indices, including hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields, often providing more interpretable models and avoiding singularities at atomic positions [25] [26].
The fundamental strength of these 3D-QSAR techniques lies in their ability to translate computed interaction fields into visual contour maps. These maps offer medicinal chemists intuitive guidance for molecular optimization by highlighting regions where modifying steric bulk or electronic characteristics would likely enhance biological activity. The application of these methods has proven valuable across various therapeutic areas, from designing novel Bcr-Abl inhibitors for chronic myeloid leukemia to developing anti-Alzheimer drug candidates targeting butyrylcholinesterase [26] [10]. This document provides a comprehensive overview of essential software tools and detailed experimental protocols to facilitate robust 3D-QSAR studies, framed within the context of advanced computational drug discovery research.
Essential Software Toolkit for 3D-QSAR Research
The successful execution of 3D-QSAR studies relies on a suite of specialized software tools, each offering distinct capabilities ranging from molecular modeling and alignment to statistical analysis and visualization. The following table summarizes the core software platforms integral to 3D-QSAR workflows.
Table 1: Essential Software Tools for 3D-QSAR Studies
| Software Tool | Primary Use in 3D-QSAR | Key Features | Licensing Model |
|---|---|---|---|
| SYBYL/X [16] [27] | Core CoMFA/CoMSIA modeling | Industry-standard for 3D-QSAR; includes molecular docking, QSAR modeling, and advanced visualization. | Commercial |
| Schrödinger Suite [28] [27] | Integrated drug discovery platform | Combines quantum mechanics, molecular dynamics, and machine learning (e.g., DeepAutoQSAR). | Modular Commercial |
| MOE (Molecular Operating Environment) [28] [27] | Comprehensive molecular modeling | Integrates cheminformatics, bioinformatics, QSAR, and structure-based design in a single package. | Commercial |
| Open3DQSAR [27] | 3D-QSAR analysis | Open-source tool dedicated to 3D-QSAR analysis, offering transparency in analytical processes. | Open-Source |
| RDKit [29] | Cheminformatics and descriptor calculation | Open-source toolkit for cheminformatics; computes molecular descriptors and fingerprints for QSAR. | Open-Source |
| StarDrop [28] [30] | AI-guided lead optimization | Platform for small molecule design and optimization with robust QSAR models for ADME properties. | Commercial |
| DataWarrior [28] | Data analysis and visualization | Open-source program combining chemical intelligence with dynamic graphical views for data analysis. | Open-Source |
| QSAR Toolbox [31] | Data gap filling and profiling | Free software for chemical hazard assessment, profiling, and read-across; incorporates numerous databases. | Free |
Beyond these specialized tools, general-purpose molecular modeling software like HyperChem is frequently used for initial geometry optimization of molecular structures [32]. Furthermore, scripting languages like Python, particularly when using libraries such as scikit-learn and pandas in conjunction with RDKit, provide a flexible environment for building custom QSAR models and automating workflows [30] [29]. The choice of software often depends on the specific research objectives, with commercial suites like Schrödinger and MOE offering all-in-one solutions with support, while open-source tools provide greater flexibility and transparency for method development.
Research Reagent Solutions: Essential Materials for 3D-QSAR
A successful 3D-QSAR study requires both software and a foundation of conceptual "research reagents" – the core components and data that form the basis of any computational model.
Table 2: Essential Research Reagents and Materials for 3D-QSAR Studies
| Reagent/Material | Function in 3D-QSAR Workflow |
|---|---|
| Curated Chemical Dataset [32] [10] | A set of molecules with consistent experimental biological activity data (e.g., IC50, Ki). This is the fundamental input for model training and validation. |
| Molecular Descriptors [30] [29] | Numerical representations of molecular structures (e.g., physicochemical properties, topological indices). RDKit is a primary tool for their calculation. |
| Profilers & Alerts (QSAR Toolbox) [31] | Pre-defined chemical functional groups or mechanistic alerts used to categorize chemicals and support read-across from data-rich analogues. |
| Force Fields (e.g., Tripos Force Field) [16] | A set of equations and parameters for calculating the potential energy of a molecular system, used for energy minimization of 3D structures. |
| Partial Least Squares (PLS) Algorithm [16] [32] | The core statistical method used to correlate the many grid-point variables (X) with the biological activity data (Y) in CoMFA/CoMSIA. |
Detailed 3D-QSAR Experimental Protocol: A CoMFA/CoMSIA Case Study
The following protocol outlines a standard workflow for conducting CoMFA and CoMSIA studies, synthesizing methodologies from several recent research applications [16] [32] [26].
Molecular Structure Preparation and Optimization
- Sketching and Initial Minimization: Sketch the 3D structures of all molecules in the dataset using a molecular modeling environment like MOE or SYBYL. Subsequently, perform energy minimization using an appropriate force field (e.g., Tripos Force Field) with a convergence criterion of 0.001 kcal/mol·Å [16].
- Charge Calculation: Calculate partial atomic charges using a method such as Gasteiger-Hückel, which is efficient for large datasets and commonly used in 3D-QSAR studies [16].
Molecular Alignment
- Template Selection: Identify a suitably rigid and highly active molecule from the dataset to serve as the template for superposition.
- Alignment Execution: Superimpose all molecules onto the template structure. Common techniques include:
- Alignment Optimization (Optional): For improved model quality, an All Orientation Search (AOS) can be performed to systematically rotate the aligned aggregate and select the orientation that yields the highest cross-validated correlation coefficient (q²) [16].
CoMFA and CoMSIA Field Calculation
- Grid Box Setup: Place the aligned molecules at the center of a 3D grid box with a spacing of 2.0 Å. The grid dimensions should extend approximately 4-5 Å beyond the union volume of all aligned molecules in every direction [16] [25].
- CoMFA Field Calculation: Using an sp³ carbon atom with a +1.0 charge as a probe, calculate steric (Lennard-Jones potential) and electrostatic (Coulombic potential) interaction energies at each grid point. Set a cutoff value of 30 kcal/mol for both fields [16].
- CoMSIA Field Calculation: Calculate similarity indices using a common probe atom for five fields: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor. Use an attenuation factor of 0.3 for the Gaussian-type distance dependence [16] [25].
Partial Least Squares (PLS) Analysis and Model Validation
- Data Filtering: Apply a column filtering value (e.g., 2.0 kcal/mol, or a lower value like 0.3 kcal/mol for more stringent noise reduction) to improve the signal-to-noise ratio by removing low-variance columns [16].
- Training/Test Set Split: Divide the dataset into a training set (typically 80-90%) for model building and a test set (10-20%) for external validation of the model's predictive power [30].
- PLS Regression: Perform PLS regression to derive the linear relationship between the CoMFA/CoMSIA descriptor fields and the biological activity values (e.g., pIC50). The optimal number of components is determined by the highest cross-validated q².
- Model Validation: Assess model quality using several statistical parameters:
- q²: The cross-validated correlation coefficient (should be >0.5 for a predictive model).
- r²: The non-cross-validated correlation coefficient for the training set.
- R²pred: The predictive r² for the external test set, calculated from the test set molecules that were excluded from model building [26] [10].
- Contour Map Generation: Visualize the results by generating 3D contour maps. These maps show regions where specific molecular fields (e.g., steric bulk, electropositive charge) are favorably or unfavorably correlated with biological activity, providing a visual guide for molecular design [10].
Figure 1: 3D-QSAR CoMFA/CoMSIA Experimental Workflow. This diagram outlines the key stages in a standard 3D-QSAR study, from initial molecular preparation to the final design of new compounds.
Integrated Workflows: Combining 3D-QSAR with Molecular Docking and Dynamics
Modern 3D-QSAR studies are increasingly integrated with other computational techniques to enhance the reliability and structural context of the models. A common and powerful strategy involves using molecular docking to define the alignment rule for 3D-QSAR [32] [10].
- Protein Preparation: Obtain the 3D crystallographic structure of the target protein from the Protein Data Bank (PDB). Add hydrogen atoms, assign partial charges, and remove water molecules except those critically involved in ligand binding [32].
- Molecular Docking: Dock all molecules in the dataset into the protein's active site using software such as AutoDock Vina or GLIDE (from Schrödinger) to generate bio-active conformations [32] [10]. This provides a structure-based alignment that may be more biologically relevant than ligand-based methods.
- Consensus Modeling: Develop multiple 3D-QSAR models using different alignment methods (e.g., docking-based, pharmacophore-based, common scaffold-based) and statistically select the best model for predictive design [32].
- Molecular Dynamics (MD) Validation: To account for protein flexibility and validate the stability of docked poses, run molecular dynamics simulations (e.g., 50-100 ns) on key ligand-protein complexes using software like GROMACS or Desmond. This step helps confirm that the binding mode used for alignment is stable over time [26] [10].
Figure 2: Integrated 3D-QSAR and Molecular Docking Workflow. This integrated approach uses molecular docking to define the bio-active conformation for alignment, resulting in more structurally-informed 3D-QSAR models that can be further validated with molecular dynamics.
Step-by-Step Protocols for CoMFA and CoMSIA Model Development
Within the framework of 3D Quantitative Structure-Activity Relationship (3D-QSAR) studies, specifically those utilizing Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), the initial assembly of a high-quality dataset is the most critical step upon which all subsequent analysis depends [11]. This protocol details the comprehensive process of assembling a congeneric series of compounds—a set of structurally related molecules that share a common core scaffold but differ in specific substituents [33]. The objective is to curate a dataset that enables the reliable construction of 3D-QSAR models capable of accurately predicting biological activity and informing the rational design of novel therapeutic agents.
Application Notes: Core Principles for a Congeneric Series
A congeneric series is fundamental to 3D-QSAR because these methods operate on the fundamental principle that all modeled compounds share a common binding mode with the biological target [33]. The following notes outline the essential criteria for the dataset:
- Common Mechanism of Action: All compounds must act via the same mechanism and have an identical or equivalent mode of binding to the target protein [33].
- Structural Congenericity: Molecules should be structurally related, typically featuring a common central scaffold or framework with variations at specific side chains or functional groups. An example from recent literature includes a series of 2,6,9-trisubstituted purine derivatives designed as Bcr-Abl inhibitors [10].
- Data Quality and Uniformity: Biological activity data (e.g., IC₅₀, Kᵢ) for all compounds must be determined using standardized and uniform protocols, preferably within a single laboratory, to minimize experimental noise and systematic bias [33] [11]. The use of inhibition constants (Kᵢ) is preferred over IC₅₀ values as they are independent of substrate concentration [33].
- Activity Range and Distribution: The biological response values should span a range as large as possible (ideally several orders of magnitude) while maintaining a symmetrical distribution around the mean to ensure a robust and predictive model [33].
Protocol: Assembling the Compound Series
Data Collection and Curation
Objective: To gather and vet chemical structures and their corresponding biological activities. Materials: Access to chemical databases (e.g., PubChem, ChEMBL, internal corporate databases), scientific literature, and experimental records.
- Compound Sourcing: Identify candidate compounds from public databases, peer-reviewed publications, or internal historical data. For the purine-based Bcr-Abl inhibitor study, a database of 58 purines was used to construct the initial models [10].
- Activity Data Compilation: Collect biological activity data for every compound. Ensure all data points are expressed in the same units and are derived from comparable assays.
- Data Filtering: Apply strict criteria to exclude compounds with ambiguous structural information or activity data obtained from non-uniform assay conditions.
Table 1: Criteria for Biological Activity Data in 3D-QSAR
| Criterion | Requirement | Rationale |
|---|---|---|
| Activity Type | Kᵢ (preferred) or IC₅₀ | Kᵢ is a direct measure of binding affinity independent of assay conditions [33]. |
| Assay Uniformity | Single source (organism/tissue/cell/protein) and laboratory | Minimizes inter-assay variability and systemic bias [33] [11]. |
| Activity Range | At least 3-4 orders of magnitude | Ensures the model captures a wide spectrum of structure-activity relationships [33]. |
| Data Distribution | Symmetrical around the mean | Prevents model skewing and overfitting to a specific activity range [33]. |
Molecular Modeling and 3D Structure Generation
Objective: To generate accurate, energy-minimized three-dimensional structures for each compound in the dataset. Materials: Cheminformatics software (e.g., RDKit, Sybyl, Schrödinger Suite).
- 2D to 3D Conversion: Convert the 2D molecular representation (e.g., SMILES strings) into a preliminary 3D structure using tools like RDKit's
AllChem.ConstrainedEmbed()or similar functions in commercial packages [11]. - Geometry Optimization: Refine the initial 3D geometry by performing energy minimization. This can be achieved using:
- Molecular Mechanics (MM): Fast and suitable for large molecules; uses force fields like UFF or MMFF [11].
- Semi-Empirical Quantum Mechanics (QM): Methods like PM3 or AM1 offer a balance between speed and accuracy for electronic property calculation [33].
- Ab Initio QM: Highly accurate but computationally intensive; recommended for final optimization of key compounds [33].
Conformational Analysis and Bioactive Conformer Selection
Objective: To identify the low-energy conformation that represents the likely bound state of the ligand to the target protein. Materials: Molecular modeling software with conformational search capabilities.
Several search methods can be employed, each with distinct advantages [33]:
- Systematic Search: Rotates all rotatable bonds by fixed increments; exhaustive but computationally demanding.
- Monte Carlo: Makes random changes to torsional angles, accepting or rejecting based on energy criteria.
- Molecular Dynamics (MD): Simulates the physical movements of atoms over time, effectively sampling the conformational landscape [34].
- Genetic Algorithm: Evolves populations of conformers based on a fitness function (e.g., low energy).
The bioactive conformation can be determined through experimental or theoretical means [33]:
- Experimental (Gold Standard): Use a 3D structure from a protein-ligand complex obtained via X-ray crystallography or NMR spectroscopy.
- Theoretical (If no structure available): Perform a conformational search and select the lowest energy conformation or use a pharmacophore-based alignment strategy. For lead optimization, a template-based method can be highly effective, using the crystal structure of a lead compound as a template to generate conformations for analogs [35].
Molecular Alignment
Objective: To superimpose all molecules in a shared 3D coordinate system that reflects their putative binding mode. Materials: Modeling software with alignment functions (e.g., MOE, Sybyl, Schrödinger).
Alignment is a critical, alignment-dependent step for CoMFA. The chosen strategy depends on available structural information [33] [11]:
- Atom-Based Alignment: Superimpose molecules based on a one-to-one pairing of atoms in a common substructure.
- Pharmacophore-Based Alignment: Align molecules according to shared pharmacophoric features (e.g., hydrogen bond donors/acceptors, hydrophobic centers).
- Database Alignment: Use the crystal structure of a lead compound as a template. Software like SkeleDock can then dock congeneric series by aligning the common scaffold of new molecules to the template, freezing these atoms, and optimizing the remaining substituents [36].
Table 2: Common Molecular Alignment Techniques
| Technique | Methodology | Use Case |
|---|---|---|
| Maximum Common Substructure (MCS) | Identifies the largest substructure shared among all molecules and uses it for superimposition [11]. | Ideal for datasets with a clearly defined and shared core scaffold. |
| Pharmacophore Alignment | Aligns molecules based on a set of abstract chemical features rather than specific atoms. | Suitable for series with significant scaffold hops but shared interaction features. |
| Template-Based (Docking) | Uses a known bioactive conformation (from X-ray) as a template for aligning other molecules [36]. | The preferred method when a high-resolution protein-ligand complex is available. |
Dataset Validation and Preparation for 3D-QSAR
Objective: To ensure the final, aligned dataset is suitable for 3D-QSAR analysis. Materials: The aligned molecular dataset; chemical space visualization tools (e.g., MolCompass [37]).
- Chemical Space Visualization: Project the final dataset into a 2D chemical space using a tool like MolCompass, which employs a parametric t-SNE model to cluster structurally similar compounds together [37]. This visual check helps confirm that the congeneric series forms a coherent cluster and identifies any potential outliers.
- Training/Test Set Division: Split the dataset into training and test sets. This can be done randomly or based on a strategic method (e.g., Kennard-Stone) to ensure the test set is representative of the chemical and activity space covered by the training set.
The following workflow diagram summarizes the entire protocol from data collection to final model readiness.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials and Software for Assembling a Congeneric Series
| Item / Reagent / Software | Function / Application in Protocol |
|---|---|
| Public Chemical Databases (e.g., PubChem, ChEMBL) | Source for chemical structures and associated bioactivity data. |
| Internal Compound Databases | Repository of proprietary compounds and assay data. |
| Cheminformatics Toolkits (e.g., RDKit, Open Babel) | Open-source libraries for 2D/3D structure manipulation, descriptor calculation, and maximum common substructure (MCS) identification [11]. |
| Commercial Modeling Suites (e.g., Schrödinger, MOE, OpenEye) | Integrated platforms for advanced molecular modeling, energy minimization, conformational search, and molecular alignment. |
| Protein Data Bank (PDB) | Primary source for experimentally determined 3D structures of proteins and protein-ligand complexes to guide bioactive conformation selection and template-based alignment [33]. |
| Visualization & Validation Tools (e.g., MolCompass) | Tools for visualizing chemical space to validate dataset consistency and model applicability domain [37]. |
| Congeneric Series of Compounds | The core set of structurally related small molecules, typically sharing a common scaffold, that is the subject of the 3D-QSAR study [10]. |
The accuracy of three-dimensional quantitative structure-activity relationship (3D-QSAR) studies, including Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), is fundamentally dependent on the quality and reliability of the initial molecular structures. Generating and optimizing 3D structures represents the critical first step in these computational workflows, establishing the foundation upon which all subsequent analyses are built. Proper 3D structure preparation ensures that the conformational sampling and molecular alignments—key components of 3D-QSAR—accurately reflect the biologically relevant orientations of molecules under investigation.
Molecular modeling techniques have become indispensable in modern drug discovery, providing powerful tools for predicting biological activity and guiding the rational design of novel therapeutic agents. The process begins with the creation of realistic 3D molecular models that serve as input for advanced computational analyses. Within the context of 3D-QSAR protocols, the generation of accurate initial structures directly influences the predictive capability of the resulting models, making this preliminary phase essential for successful outcomes in computer-aided drug design campaigns targeting various disease pathways, including oncology, metabolic disorders, and infectious diseases.
Practical Applications in Drug Discovery
The application of robust 3D structure generation protocols has demonstrated significant value across multiple therapeutic areas, enabling the identification and optimization of novel chemical entities with improved target affinity and selectivity.
Table 1: Recent Applications of 3D-QSAR and Molecular Modeling in Drug Discovery
| Therapeutic Area | Target Protein | Modeling Approaches | Key Outcomes | Citation |
|---|---|---|---|---|
| Oncology | Tyrosine Threonine Kinase (TTK) | 3D-QSAR, Molecular Docking, MD Simulations | Designed novel compounds with predicted improved activity; models showed q² = 0.583-0.690 | [38] |
| Oncology | Bcr-Abl | 3D-QSAR, CoMFA, CoMSIA | New purine derivatives with IC₅₀ = 0.13-0.19 μM surpassed imatinib potency | [10] |
| Endocrinology | Estrogen Receptor Alpha (ERα) | Machine Learning-based 3D-QSAR | MLP 3D-QSAR model outperformed conventional VEGA models in accuracy and sensitivity | [5] |
| Metabolic Disease | α-Glucosidase | CoMFA, CoMSIA, Molecular Docking | Developed models with Q² = 0.600-0.616 and R² = 0.928-0.958; designed four new potent inhibitors | [39] |
| Oncology | VEGFR-2 | 3D-QSAR, CoMFA, CoMSIA, MD | Established models with R²cv = 0.663 (CoMFA) and R²pred = 0.6974 (CoMSIA) | [40] |
| Infectious Disease | β-haematin | CoMFA, CoMSIA, HQSAR | Prioritized 125 indolo[3,2-c] quinolone analogues as potential antimalarials | [41] |
Experimental Protocols
Protocol 1: Initial Structure Generation and Geometry Optimization
The process of generating biologically relevant 3D molecular structures begins with careful construction and optimization of molecular geometry.
Structure Sketching and Initial Geometry
- Draw two-dimensional molecular structures using molecular editing software such as Maestro's 2D builder [38].
- Convert 2D structures to 3D representations using standard conversion algorithms that establish appropriate bond lengths, angles, and torsions.
- For novel compounds not available in structural databases, molecular mechanics methods (e.g., MMFF94) can generate initial coordinates based on established force field parameters.
Geometry Optimization and Partial Charge Calculation
- Perform initial energy minimization using conjugate gradient and Powell methods to relieve severe steric clashes and achieve reasonable starting geometry [38].
- Calculate partial atomic charges using appropriate methods:
- Gasteiger-Hückel: Efficient for large datasets with reasonable accuracy for electrostatic field calculations [38].
- Merck Molecular Force Field (MMFF94): Provides excellent performance in 3D-QSAR studies, particularly for diverse compound sets [38].
- Pullman Methods: Alternative approach for charge calculation in specific molecular systems.
- Compare charge models to identify the most appropriate for the specific molecular series under investigation.
Conformational Analysis
- Generate representative conformational ensembles using systematic search, stochastic methods, or molecular dynamics simulations.
- Select the lowest energy conformation or biologically relevant conformation (if known from structural biology data) for alignment and analysis.
- For flexible molecules, consider multiple low-energy conformers to assess the impact of conformational flexibility on model quality.
Protocol 2: Molecular Dynamics Simulations for Conformational Sampling and Validation
Molecular dynamics (MD) simulations provide a powerful approach for sampling conformational space and validating the stability of ligand-receptor complexes.
System Setup
- Obtain initial protein coordinates from the Protein Data Bank (PDB) or generate through homology modeling if experimental structures are unavailable [42].
- Process the initial structure using
pdb2gmxcommand in GROMACS to generate molecular topology and coordinate files in GROMACS format (.gro) [42]. - Select an appropriate force field (e.g., ffG53A7 for proteins with explicit solvent) when prompted during the
pdb2gmxexecution [42].
Simulation Environment Preparation
- Define the simulation volume using
editconfto create a periodic boundary box (cubic, dodecahedron, or octahedron) with a minimum distance of 1.4 nm between the protein and box edge [42]. - Solvate the system using the
solvatecommand, which adds explicit water molecules (e.g., SPC, TIP3P, TIP4P models) to the simulation box [42]. - Neutralize the system charge using the
genioncommand by adding appropriate counterions (e.g., Na⁺, Cl⁻) to achieve overall charge neutrality [42].
- Define the simulation volume using
Energy Minimization and Equilibration
- Perform energy minimization using the steepest descent or conjugate gradient algorithm to remove steric clashes and unfavorable contacts.
- Conduct equilibration in two phases: (1) NVT ensemble to stabilize temperature, and (2) NPT ensemble to stabilize pressure and density.
- Generate the run input file using the
gromppcommand, which collects parameters, topology, and coordinates into a single binary file (.tpr) [42].
Production MD and Analysis
- Execute production MD simulations for time scales appropriate to the biological process of interest (typically nanoseconds to microseconds).
- Analyze trajectories using GROMACS tools or custom scripts to calculate root-mean-square deviation (RMSD), radius of gyration (Rg), hydrogen bonding patterns, and other relevant structural metrics.
- Perform molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) calculations to estimate binding free energies for protein-ligand complexes [38].
Diagram 1: Workflow for 3D Structure Generation and Modeling in Drug Discovery. This diagram illustrates the integrated protocol for generating and optimizing 3D molecular structures, culminating in their application for 3D-QSAR modeling and structure-based drug design.
Successful implementation of 3D structure generation and optimization protocols requires access to specialized software tools, computational resources, and methodological frameworks.
Table 2: Essential Computational Tools for 3D Structure Generation and Modeling
| Tool/Resource | Type | Primary Function | Application in 3D-QSAR |
|---|---|---|---|
| Maestro | Graphical Interface | Molecular visualization, structure building, and project management | 2D to 3D structure conversion, molecular editing, and visualization of results [38] |
| GROMACS | Molecular Dynamics Suite | MD simulations, energy minimization, and trajectory analysis | Conformational sampling, validation of structural stability, and binding free energy calculations [43] [42] |
| SYBYL | Molecular Modeling | CoMFA and CoMSIA field calculations, molecular alignment | 3D-QSAR model development using steric, electrostatic, and hydrophobic fields [38] |
| Schrödinger Suite | Comprehensive Drug Discovery Platform | Protein preparation, molecular docking, FEP+ calculations, QM workflows | Structure preparation, binding mode prediction, and free energy calculations [44] |
| RasMol | Molecular Visualization | Structure visualization and rendering | Inspection of protein structures and graphics rendering [42] |
| Merck Molecular Force Field (MMFF94) | Force Field | Molecular mechanics calculations | Partial charge calculation and geometry optimization [38] |
| Protein Data Bank (PDB) | Structural Database | Repository of experimentally-determined structures | Source of initial protein coordinates for structure-based design [42] |
Integrated Workflow for 3D-QSAR Model Development
The integration of 3D structure generation with subsequent computational analyses creates a powerful pipeline for rational drug design.
Molecular Alignment and Field Calculation
Molecular alignment represents a critical step in 3D-QSAR studies, directly influencing model quality and interpretability.
Alignment Strategies
- Ligand-based alignment: Use the most active compound as a template for superimposing all molecules in the dataset based on common structural features [38].
- Structure-based alignment: Align molecules according to their predicted binding orientations within the target protein's active site [38].
- Docking-based alignment: Employ molecular docking to generate putative binding modes and use these for molecular superposition.
CoMFA and CoMSIA Field Calculations
- Establish a 3D grid with appropriate spacing (typically 2.0 Å) encompassing all aligned molecules [38].
- Calculate steric (Lennard-Jones) and electrostatic (Coulombic) potential fields for CoMFA using a sp³ carbon atom with +1.0 charge as the probe [38].
- For CoMSIA, compute additional similarity fields including hydrophobic, hydrogen bond donor, and hydrogen bond acceptor properties [39].
- Apply an energy cutoff of 30 kcal/mol to truncate extreme values and prevent model dominance by a few high-energy interactions [38].
Model Validation and Application
Robust validation ensures the reliability and predictive capability of developed 3D-QSAR models.
Statistical Validation
- Apply leave-one-out (LOO) cross-validation to calculate q² values, with q² > 0.5 indicating good predictive ability [10] [38].
- Use non-cross-validation to compute conventional r² values, assessing the model's capability to explain variance in the training set.
- Evaluate model robustness through bootstrapping analysis (typically 100 runs) [38].
- Assess external predictive ability using an independent test set of compounds not included in model development.
Model Interpretation and Compound Design
- Interpret contour maps to identify regions where specific molecular properties enhance or diminish biological activity.
- Design novel compounds incorporating structural features predicted to improve potency based on contour map analysis [39] [38].
- Synthesize and experimentally validate top-predicted compounds to confirm model predictions and refine subsequent iterations.
Diagram 2: 3D-QSAR Model Development and Validation Workflow. This diagram outlines the key steps in developing predictive 3D-QSAR models following the generation and optimization of 3D molecular structures.
The generation and optimization of 3D molecular structures represents a fundamental process in computational drug discovery, serving as the critical foundation for successful 3D-QSAR studies. Through the systematic application of the protocols outlined in this application note—encompassing initial structure generation, conformational analysis, molecular dynamics simulations, and rigorous validation procedures—researchers can establish reliable computational models with demonstrated predictive capability across multiple therapeutic areas. The integrated workflow combining 3D-QSAR with complementary structure-based approaches continues to provide valuable insights for rational drug design, significantly reducing the time and resources required to advance promising compounds through the discovery pipeline. As computational methods continue to evolve, particularly through the incorporation of machine learning and advanced sampling techniques, the precision and applicability of structure-based modeling approaches will further expand, enhancing their role as indispensable tools in modern drug development.
In the realm of three-dimensional quantitative structure-activity relationship (3D-QSAR) studies, including Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), molecular alignment stands as the most critical and sensitive step determining model success or failure [45]. The fundamental principle of 3D-QSAR relies on comparing molecular interaction fields surrounding a set of compounds, and this comparison becomes statistically meaningful only when molecules are aligned in a biologically relevant manner [2]. Unlike 2D-QSAR methods that utilize fixed molecular descriptors, 3D-QSAR inputs are dependent on the relative orientation and conformation of molecules in space, making alignment quality the primary source of both signal and noise in the resulting models [45].
The biological receptor perceives a ligand not as a set of atoms and bonds, but as a shape carrying complex electrostatic and steric forces [2]. Molecular alignment aims to replicate this biological perception by superimposing molecules in a way that mimics their binding orientation within the target protein's active site. When executed correctly, proper alignment enables 3D-QSAR to reveal subtle structural determinants of biological activity; when performed poorly, it generates models with limited predictive power that may lead to erroneous structural insights [45].
Molecular Alignment Fundamentals
Theoretical Basis for Alignment in 3D-QSAR
Molecular alignment for 3D-QSAR is fundamentally based on the concept that bioactive molecules share common interaction patterns with their biological target, even when their chemical scaffolds differ. The alignment process positions molecules in three-dimensional space to maximize the overlap of these potential interaction points. In field-based methods, molecules are aligned according to their molecular electrostatic potentials and steric fields, which represent how the receptor would "see" the ligands [2].
The importance of alignment stems from its direct impact on the calculation of molecular interaction fields (MIFs). MIFs are measured by placing probe atoms at grid points surrounding the aligned molecules and calculating interaction energies using potential functions such as Coulomb's law for electrostatic fields and Lennard-Jones potentials for steric fields [2]. Even minor misalignments can significantly alter these field values, consequently affecting the statistical model derived from them. As noted in one analysis, "If your alignments are incorrect your model will have limited or no predictive power" [45].
Comparison of Alignment-Dependent and Alignment-Independent Approaches
| Feature | Alignment-Dependent Methods | Alignment-Independent Methods |
|---|---|---|
| Core Principle | Direct superposition of molecules in 3D space | Conversion of 3D properties into alignment-independent descriptors |
| Key Methods | CoMFA, CoMSIA [46] [47] | GRIND, 3D-QSDAR [48] [49] |
| Descriptor Type | Grid-based interaction fields [2] | GRID Independent Descriptors (GRIND) [49] |
| Handling of Flexibility | Requires conformation selection | Often uses multiple conformations |
| Interpretability | Direct visual interpretation of contour maps [47] | Requires additional steps for structural interpretation |
| Computational Demand | High (alignment-critical) | Lower (automated) |
| Primary Challenge | Determining biologically relevant alignment | Preserving 3D spatial information |
Molecular Alignment Strategies and Methodologies
Structure-Based Alignment
Structure-based alignment utilizes known structural information from the target protein, typically from X-ray crystallography or homology models, to guide molecular superposition. In this approach, molecules are docked into the protein's binding site, and their poses are used as the basis for alignment [38]. This method provides a biologically relevant framework for alignment, as it directly reflects how molecules position themselves when interacting with the target.
The protocol for structure-based alignment typically involves:
- Protein Preparation: Obtain and prepare the protein structure by removing water molecules, adding hydrogen atoms, and optimizing hydrogen bonding networks [47].
- Ligand Docking: Dock each ligand into the binding site using molecular docking software such as AutoDock or Glide [47] [38].
- Pose Extraction: Extract the lowest energy or most representative docking pose for each compound.
- Reference Selection: Choose a reference molecule, typically a high-affinity ligand with a known binding mode.
- Superposition: Align all molecules to the reference based on their docked positions [38].
A study on TTK inhibitors demonstrated the effectiveness of this approach, where "structure-based alignment yielded highly predictive CoMFA (q² = 0.583, Predr² = 0.751) and CoMSIA (q² = 0.690, Predr² = 0.767) models" [38].
Ligand-Based Alignment
Ligand-based alignment strategies rely solely on the properties of the ligands themselves, making them particularly valuable when structural information about the target protein is unavailable. The most common ligand-based approaches include:
- Pharmacophore-Based Alignment: Molecules are aligned to maximize the overlap of key pharmacophoric features such as hydrogen bond donors/acceptors, hydrophobic centers, and charged groups [50].
- Field-Based Alignment: Molecules are aligned according to their similarity in molecular electrostatic potential and steric fields, often using algorithms such as FBSS (Field-Based Similarity Searching) [50].
- Common Substructure Alignment: A shared structural scaffold is identified and used as the basis for molecular superposition [45].
The protocol for common substructure alignment includes:
- Identify Common Core: Determine the maximum common substructure shared across the data set.
- Template Selection: Choose a reference molecule with representative structure and activity.
- Structural Alignment: Superimpose all molecules onto the template based on the common substructure atoms.
- Field Optimization: Refine alignments using field similarity metrics to optimize electrostatic and steric overlap [45].
A critical consideration in ligand-based alignment is handling molecules that extend beyond the common core. As noted by experts, "For most data sets I find that you need 3-4 reference molecules to fully constrain all of the others" [45].
Automated and Field-Based Alignment Methods
Automated alignment methods aim to reduce the subjectivity and time investment required for manual molecular alignment. These approaches use computational algorithms to generate consistent alignments based on objective criteria. The FBSS (Field-Based Similarity Searching) method represents one such automated approach that uses molecular field similarity to generate alignments [50].
Research comparing automated versus manual alignments has shown that "the QSAR models resulting from the FBSS alignments are broadly comparable in predictive performance with the models resulting from manual alignments" [50]. This suggests that automated methods can provide a valuable starting point for 3D-QSAR analyses, particularly for large datasets where manual alignment would be prohibitively time-consuming.
The FBSS methodology operates by:
- Field Calculation: Computing steric and electrostatic fields for each molecule.
- Similarity Scoring: Calculating field similarity scores between pairs of molecules.
- Alignment Optimization: Iteratively optimizing molecular orientations to maximize field similarity.
- Consensus Building: Generating a consensus alignment that maximizes overall field similarity across the dataset [50].
Alignment-Independent 3D-QSAR Methods
For situations where molecular alignment proves particularly challenging, alignment-independent 3D-QSAR methods offer an alternative approach. These techniques transform 3D molecular information into descriptors that do not require molecular superposition, thereby bypassing alignment-related issues [48] [49].
The GRIND (GRID Independent Descriptor) methodology exemplifies this approach:
- MIF Calculation: Compute Molecular Interaction Fields using various GRID probes.
- Descriptor Extraction: Extract and encode the most relevant regions of interaction fields into alignment-independent variables.
- Model Building: Use these descriptors as inputs for statistical model development [49].
Another alignment-independent technique, 3D-QSDAR, uses a different approach based on "a unique 'fingerprint' constructed from the NMR chemical shifts, δ, of all carbon atom pairs placed on the X- and Y-axes joined with the inter-atomic distances between each pair on the Z-axis" [48]. This method has demonstrated comparable predictive ability to alignment-dependent methods while requiring significantly less computational time [48].
Molecular Alignment Decision Workflow: This diagram illustrates the strategic decision process for selecting appropriate molecular alignment methods in 3D-QSAR studies, highlighting the critical branching point based on protein structural information availability.
Practical Protocols for Molecular Alignment
Comprehensive Alignment Protocol for 3D-QSAR Studies
A robust alignment protocol should combine multiple approaches to achieve biologically meaningful results. The following step-by-step protocol integrates best practices from literature:
Initial Preparation
Reference Molecule Selection
- Identify a representative, conformationally restricted compound with high biological activity as the primary template [45].
- If available, use a ligand with a experimentally determined bioactive conformation.
Multi-Reference Alignment Strategy
- Align all compounds to the primary template using substructure alignment for the common core.
- Identify compounds with substituents extending beyond regions covered by the primary template.
- Select additional reference molecules to constrain these regions and realign the dataset [45].
- "For most data sets I find that you need 3-4 reference molecules to fully constrain all of the others" [45].
Validation and Refinement
- Visually inspect alignments for all compounds, paying attention to conservative regions.
- Ensure consistent orientation of key functional groups across the series.
- Verify that alignment choices are made without reference to biological activity data to avoid bias [45].
Final Alignment
- Generate the final aligned dataset for CoMFA/CoMSIA analysis.
- Document all alignment decisions and reference molecules used.
Common Pitfalls and Quality Control Measures
| Common Pitfall | Impact on Model | Quality Control Measure |
|---|---|---|
| Activity-Based Alignment Tweaking | Invalid, over-optimistic models [45] | Finalize alignments before viewing activity data |
| Inconsistent Partial Charges | Altered electrostatic fields [38] | Use consistent charge calculation methods across all molecules |
| Ignoring Molecular Flexibility | Non-bioactive conformations | Use multiple low-energy conformers or conformationally restricted templates |
| Over-reliance on Single Template | Poor alignment for diverse structures | Implement multi-reference alignment strategy [45] |
| Neglecting Visual Inspection | Undetected alignment errors | Systematic visual check of all molecular overlays |
A critical warning from experienced practitioners emphasizes: "You must not change the X data while paying attention (either directly or indirectly) to the Y data (the activities)" [45]. Aligning molecules based on their activity values represents a form of circular reasoning that produces statistically invalid models with exaggerated predictive metrics.
Research Reagents and Computational Tools
Essential Software Tools for Molecular Alignment
| Tool Name | Alignment Method | Key Features | Applicability |
|---|---|---|---|
| SYBYL [47] | Ligand-based, Pharmacophore | CoMFA/CoMSIA implementation, Field calculation | Comprehensive 3D-QSAR platform |
| FBSS [50] | Field-based similarity | Automated alignment, Electrostatic and steric field optimization | Large datasets, Initial screening |
| Pentacle [49] | Alignment-independent (GRIND) | GRID MIFs, GRIND descriptors | Challenging alignment scenarios |
| PharmQSAR [51] | Multiple algorithms | QM-based fields, Automated workflow | Lead optimization, Property prediction |
| Forge/Torch [45] | Field-based, Template | FieldTemplater, Multi-reference alignment | Detailed 3D-QSAR with visual analysis |
Key Computational Reagents and Parameters
Successful molecular alignment requires careful attention to computational parameters that function as "research reagents" in silico:
- Partial Charge Calculation Methods: Gasteiger-Huckel, MMFF94, AM1, or other semi-empirical methods significantly impact electrostatic field calculations and subsequent alignments [38]. Consistency across the dataset is crucial.
- Force Field Selection: Tripos Force Field, Merck Molecular Force Field (MMFF94), or other parameter sets control energy minimization and conformational sampling [47].
- Probe Atoms: Standard probes include sp³ carbon with +1 charge for electrostatic fields and neutral sp³ carbon for steric fields in CoMFA [2].
- Grid Parameters: Typical spacing of 1-2 Å between grid points, with extension beyond molecular dimensions by 4 Å in all directions [47].
- Energy Thresholds: Steric and electrostatic field energy values are typically truncated at 30 kcal/mol to prevent extreme values from dominating the analysis [47].
Molecular alignment remains the cornerstone of successful 3D-QSAR studies, with the alignment strategy directly determining the quality and predictive power of resulting models. The fundamental principle reiterated across literature is that "the majority of the signal is in the alignments" [45]. As 3D-QSAR methodologies continue to evolve, incorporating machine learning approaches [5] and advanced field-based alignment algorithms, the critical importance of biologically relevant molecular superposition remains unchanged.
Researchers must select alignment strategies based on available structural information, chemical diversity of the dataset, and the specific biological context. Structure-based alignment provides the most direct link to biological reality when protein structural information is available, while sophisticated ligand-based approaches offer viable alternatives when such information is lacking. Regardless of the specific method chosen, the alignment process must be performed meticulously, with careful attention to potential pitfalls and strict avoidance of activity-based bias.
When executed with scientific rigor, proper molecular alignment enables 3D-QSAR to reveal subtle structural determinants of biological activity, providing valuable insights for rational drug design and chemical optimization. The protocols and strategies outlined in this application note provide a framework for achieving such rigorous alignments, forming the essential foundation for meaningful 3D-QSAR analyses.
In the realm of computer-aided drug design, Quantitative Structure-Activity Relationship (QSAR) studies serve as a cornerstone for understanding the molecular basis of biological activity. Three-dimensional QSAR (3D-QSAR) techniques, notably Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), extend traditional QSAR by incorporating spatial and field-based molecular descriptors [52] [17]. These methodologies correlate the steric, electrostatic, and hydrophobic fields surrounding a set of molecules with their measured biological potency, enabling the prediction of new compounds and providing visual insights for rational drug design. This application note details the theoretical underpinnings, calculation protocols, and practical applications of these critical molecular field descriptors within the context of 3D-QSAR CoMFA/CoMSIA research for drug development professionals.
Theoretical Background of Molecular Fields
Molecular fields are 3D representations of physicochemical properties that dictate how a ligand interacts with its biological target. The core premise is that the binding affinity and selectivity of a molecule are determined by the complementarity of its interaction fields with the receptor's binding site.
Figure 1: Computational workflow for deriving 3D-QSAR models from molecular field calculations. Aligned molecular structures are used to compute steric, electrostatic, and hydrophobic fields, which are then correlated with biological activity to generate predictive models.
Fundamental Field Types in CoMFA and CoMSIA
The following table summarizes the key molecular fields utilized in CoMFA and CoMSIA studies.
Table 1: Core Molecular Field Descriptors in 3D-QSAR
| Field Type | Physical Basis | Role in Ligand-Target Interaction | Primary Computational Method |
|---|---|---|---|
| Steric | Molecular size and shape [53] | Governs van der Waals interactions and physical fit within the binding site; excessive steric bulk can lead to clashes, while insufficient bulk reduces contact surface area. | Lennard-Jones potential |
| Electrostatic | Distribution of positive and negative charges [53] | Dictates directional interactions such as hydrogen bonding, ion pairing, and dipole-dipole interactions, crucial for binding affinity and specificity. | Coulombic potential |
| Hydrophobic | Tendency to avoid water (logP) [54] | Drives desolvation and the association of non-polar surfaces; optimal hydrophobic complementarity enhances binding free energy. | Based on thermodynamic measurements and group contributions |
Calculation Methodologies and Protocols
Computational Workflow for Field Descriptor Calculation
The accurate calculation of molecular fields requires a structured workflow to ensure the resulting 3D-QSAR models are robust and predictive.
Figure 2: Key stages in the 3D-QSAR pipeline, highlighting the critical structure preparation and alignment phase that precedes field calculation.
Protocol for Calculating Steric and Electrostatic Fields (CoMFA-like)
This protocol outlines the steps for calculating the steric and electrostatic fields that form the basis of a CoMFA study [52].
Objective: To generate spatial descriptors of steric and electrostatic properties for a set of aligned molecules for use in 3D-QSAR analysis.
Materials/Software:
- Molecular structures of compounds with associated biological activity data (e.g., pKi, IC50).
- Computational chemistry software (e.g., Sybyl, OpenEye Toolkits, MOE, Schrödinger Suite).
- Force Field Parameterization Software: Required for energy minimization and partial charge calculation (e.g., MMFF94, AMBER-ff19SB, GAFF2) [52] [55].
- Quantum Chemistry Software (Optional but Recommended): For accurate electrostatic potential-derived charges (e.g., Gaussian, Gamess, MOPAC) [56].
Procedure:
- Structure Preparation and Energy Minimization:
- Build or import all molecular structures.
- Add hydrogens and assign correct protonation states at physiological pH (7.4).
- Assign initial partial atomic charges (e.g., Gasteiger-Hückel method) [52].
- Perform geometry optimization using a molecular mechanics force field (e.g., MMFF94) until a convergence criterion is reached (e.g., gradient < 0.05 kcal/mol·Å) [52].
Molecular Alignment (Superposition):
- This is the most critical step, as the quality of the alignment dictates the success of the 3D-QSAR model.
- Select a template molecule, typically the most active or rigid compound.
- Align all molecules based on a common scaffold or pharmacophoric features using rigid-body or flexible alignment algorithms [52].
- Ensure the alignment reflects a putative binding mode.
Calculation of Interaction Fields:
- Place the aligned molecules within a 3D grid with a default spacing of 2.0 Å, ensuring the grid extends beyond the molecular dimensions by at least 4.0 Å in all directions.
- Steric Field: At each grid point, calculate the steric interaction energy using a Lennard-Jones potential (e.g., with a sp³ carbon probe atom with a van der Waals radius of 1.52 Å and a charge of +1.0) [52].
- Electrostatic Field: At each grid point, calculate the electrostatic interaction energy using a Coulombic potential with a distance-dependent dielectric constant (e.g., ε = 1r or ε = 4r).
Data Reduction and Model Building:
- The resulting field values at thousands of grid points serve as descriptors.
- Use Partial Least Squares (PLS) regression to correlate the field descriptors with the biological activity data, employing cross-validation (e.g., leave-one-out) to determine the optimal number of components and prevent overfitting [52] [57].
Protocol for Calculating CoMSIA Fields
CoMSIA extends CoMFA by incorporating additional fields and using a Gaussian function to avoid singularities, leading to more interpretable models [52] [17].
Objective: To generate similarity indices descriptors based on steric, electrostatic, hydrophobic, and hydrogen-bonding properties.
Materials/Software: Same as Protocol 3.2.
Procedure:
- Follow steps 1 and 2 from the CoMFA protocol for structure preparation and alignment.
- Calculation of Similarity Indices:
- Use the same 3D grid as in CoMFA.
- For each field type, calculate the similarity of each molecule to a probe atom at every grid point using a Gaussian function. The default attenuation factor (α) is typically 0.3.
- The five CoMSIA fields include:
- Steric: Defined by the third power of the atomic radii.
- Electrostatic: Derived from partial atomic charges.
- Hydrophobic: Based on atom-based parameters (e.g., Rekker's or Viswanadhan's fragmental constants).
- Hydrogen Bond Donor
- Hydrogen Bond Acceptor
- Model Building and Validation:
- The calculated similarity indices are used as descriptors in a PLS analysis, similar to the CoMFA procedure.
Research Reagent Solutions: Computational Tools
A suite of software tools is available to computational researchers for calculating molecular descriptors and building 3D-QSAR models.
Table 2: Key Software Tools for Molecular Descriptor Calculation and 3D-QSAR
| Tool Name | Type | Primary Function in Descriptor Calculation | Application in 3D-QSAR |
|---|---|---|---|
| Sybyl | Commercial Suite | Industry-standard for CoMFA and CoMSIA analyses; provides robust molecular alignment and field calculation algorithms. | Direct implementation of CoMFA/CoMSIA [52]. |
| OpenEye 3D-QSAR | Commercial Tool | Uses descriptors based on molecular shape (ROCS) and electrostatic potential (EON) similarity for robust, interpretable models [53]. | Consensus model for binding affinity prediction [53]. |
| RDKit | Open-Source Library | Calculates a wide range of 2D and 3D molecular descriptors; facilitates structure preprocessing and manipulation. | Data preparation, descriptor generation, and cheminformatics analysis [58]. |
| Dragon | Commercial Software | Computes over 5,000 molecular descriptors, making it one of the most comprehensive descriptor calculation tools available. | Generation of extensive descriptor sets for QSAR [58]. |
| Gaussian/GAMESS | Quantum Chemistry | Performs ab initio calculations to derive highly accurate electronic properties (e.g., electrostatic potentials, HOMO/LUMO energies) [56]. | Calculation of reliable partial atomic charges for electrostatic fields. |
| MOPAC | Semi-Empirical QM | Provides a faster, approximate quantum mechanical method suitable for larger molecules, enabling calculation of properties like polarizability [56]. | Estimation of electronic descriptors for large datasets. |
Applications in Drug Discovery
The application of steric, electrostatic, and hydrophobic field calculations is instrumental in addressing key challenges in modern drug discovery. For instance, in the design of 5-HT1A receptor ligands, 3D-QSAR studies using CoMFA and CoMSIA were pivotal in elucidating the structural features governing both high affinity and selectivity over the closely related α1-adrenoreceptor [52]. The resulting models successfully correlated steric bulk in specific regions with enhanced selectivity, while electrostatic and hydrophobic fields dictated binding affinity, allowing researchers to propose novel chemical scaffolds with optimized profiles.
Similarly, in the development of phosphodiesterase 4 (PDE4) inhibitors, a 3D-QSAR pharmacophore model featuring two hydrogen-bond acceptors and two hydrophobic features was developed [17]. Subsequent CoMFA and CoMSIA models, built based on this pharmacophore alignment, exhibited high predictive power (q² > 0.75, R² > 0.96), enabling the rational design of new phenyl alkyl ketone derivatives with predicted high activity and desirable drug-like properties [17]. These case studies underscore the power of field-based descriptors in a generative design cycle, where model interpretations directly inspire new chemical ideas.
Advanced Considerations and Emerging Trends
While the calculation of standard molecular fields is well-established, several advanced considerations are crucial for success. The treatment of molecular alignment and conformation remains a primary challenge; the choice of the "bioactive" conformation and a meaningful alignment rule can significantly impact the model [52]. Furthermore, the inclusion of entropy-related descriptors, such as the number of rotatable bonds, can improve model predictivity for flexible ligands.
Emerging trends in the field are expanding the toolkit available to computational scientists. Water-based pharmacophore modeling is a promising ligand-independent approach that utilizes molecular dynamics (MD) simulations of water molecules in an empty binding site to identify interaction hotspots [55] [59]. These "water pharmacophores" can be used for virtual screening and provide complementary information to traditional field-based methods. Another advancement is the use of alignment-independent 3D-QSAR techniques, such as Comparative Molecular Moment Analysis (CoMMA), which utilizes moments of the molecular mass and charge distributions, thus eliminating the sensitive superposition step entirely [57]. Finally, the integration of machine learning algorithms with classical descriptor sets is enhancing predictive performance and feature interpretation, as demonstrated by OpenEye's use of Gaussian Process Regression and kernel PLS in their 3D-QSAR tool [53].
Building the QSAR Model Using Partial Least Squares (PLS) Regression
In the field of three-dimensional quantitative structure-activity relationship (3D-QSAR) modeling, Partial Least Squares (PLS) regression serves as the fundamental statistical engine for correlating complex molecular descriptor data with biological activity [11]. This technique is particularly indispensable for handling the high-dimensional, multicollinear, and noisy datasets generated by 3D-QSAR methodologies such as Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) [19] [12]. In CoMFA, steric (Lennard-Jones) and electrostatic (Coulombic) potential energies are calculated at thousands of grid points surrounding aligned molecules, while CoMSIA extends this to include hydrophobic, and hydrogen bond donor and acceptor fields, all using Gaussian-type functions for smoother field distributions [19] [12]. PLS regression effectively navigates this descriptor complexity by projecting the original variables into a reduced set of uncorrelated latent components that maximize the covariance between the molecular fields and the biological response variable [11] [60]. This capability makes PLS the standard algorithmic approach for establishing robust, interpretable, and predictive 3D-QSAR models in modern computational drug discovery.
Theoretical Foundation of PLS Regression
Mathematical Principles
The PLS algorithm operates by simultaneously projecting both the predictor variables (X-block, representing molecular field values) and response variables (Y-block, representing biological activities) into a common latent variable space [11] [60]. This projection is mathematically represented as X = TPᵀ + E and Y = UQᵀ + F, where T and U are the score matrices containing the latent variables for X and Y respectively, P and Q are the loading matrices that show how strongly each original variable contributes to the latent components, and E and F represent the residual matrices [60]. The algorithm maximizes the covariance between the X- and Y-score vectors, effectively filtering out noise while preserving the structurally relevant information that correlates with biological activity [11]. For 3D-QSAR applications, this means that despite thousands of potentially correlated grid points being analyzed, the model focuses only on those spatial regions where field variations systematically correspond to changes in measured activity such as IC₅₀ or Kᵢ values [12] [60].
Addressing Multicollinearity in 3D-QSAR Descriptors
The rectangular data structure inherent to 3D-QSAR presents significant statistical challenges that PLS specifically addresses. A typical CoMFA or CoMSIA study might encompass 20-50 compounds but generate thousands of steric and electrostatic field values from the grid points surrounding the aligned molecular set [11]. These field descriptors are highly correlated because interaction energies at adjacent grid points tend to be similar, creating multicollinearity that renders traditional multiple linear regression (MLR) inappropriate [11]. PLS regression circumvents this limitation by constructing latent components that are linear combinations of the original variables, with the number of components optimized through cross-validation techniques to prevent overfitting [19] [60]. This dimensional reduction capability ensures that 3D-QSAR models remain statistically valid and predictive even when the number of variables dramatically exceeds the number of observations, a common scenario in pharmaceutical research where compound synthesis is costly and time-consuming [12].
Experimental Protocol for PLS-Based 3D-QSAR
Pre-Modeling Data Preparation
The foundation of any reliable 3D-QSAR model lies in meticulous data preparation, beginning with the assembly of a congeneric compound series with uniformly determined biological activities (e.g., IC₅₀, Kᵢ, or MIC values) [11]. These activity values are typically converted to negative logarithmic scales (pIC₅₀, pKᵢ, or pMIC) to linearize the relationship with binding energy [60]. Following compound selection, the critical step of molecular alignment is performed using either common substructure approaches (e.g., Bemis-Murcko scaffolds or maximum common substructure) or pharmacophore-based methods to ensure all molecules share a consistent orientation in 3D space [11]. Tools such as GALAHAD have demonstrated superior performance for pharmacophore alignment in studies on α1A-adrenergic receptor antagonists [12]. With aligned structures, molecular descriptors are computed by placing compounds within a 3D grid (typically with 1.0-2.0 Å spacing) and calculating interaction energies between a probe atom and each molecule at every grid point [12]. For CoMFA, steric (Lennard-Jones) and electrostatic (Coulombic) fields are computed, while CoMSIA incorporates additional similarity fields including hydrophobic and hydrogen-bonding descriptors using Gaussian functions to smooth field distributions [19] [12].
Core PLS Modeling Procedure
The PLS modeling workflow begins with cross-validation to determine the optimal number of components that balance model complexity with predictive power [19] [60]. Leave-one-out (LOO) cross-validation is most commonly employed, where each compound is systematically excluded from the training set, a model is built with the remaining compounds, and the activity of the excluded compound is predicted [12] [60]. The cross-validated correlation coefficient (q²) is calculated as q² = 1 - PRESS/SS, where PRESS is the prediction error sum of squares and SS is the total sum of squares of the activity values [60]. A q² value > 0.5 is generally considered statistically significant, while q² > 0.9 indicates excellent predictive capability [12]. Following component optimization, the final PLS model is built using all training set compounds with the optimal number of components, generating conventional correlation coefficients (r²) and standard errors of estimate [19]. The model's predictive robustness is then evaluated using an external test set of compounds that were not included in model development, with the predictive r² (r²pred) providing the most stringent assessment of real-world utility [12].
Model Validation and Interpretation
Comprehensive validation represents the most critical phase in establishing a reliable 3D-QSAR model. The bootstrapping technique is frequently employed to assess the internal stability and statistical significance of the derived model by repeatedly sampling the dataset with replacement and recalculating model parameters [60]. For the final validated model, interpretation occurs primarily through visualization of coefficient contour maps that identify specific spatial regions where molecular properties favorably or unfavorably influence biological activity [11]. These maps are typically superimposed on a reference compound, with different colors indicating regions where increased steric bulk (green), decreased steric bulk (yellow), electropositive groups (blue), or electronegative groups (red) would enhance activity [11]. In the case of CoMSIA, additional maps represent hydrophobic (yellow-brown), hydrogen bond donor (cyan), and hydrogen bond acceptor (magenta) favorable regions [19] [12]. These visual representations transform complex statistical models into intuitive design guides that practicing medicinal chemists can utilize to prioritize molecular modifications for synthesis [11].
Application Case Studies
Benchmark Validation with Steroid Dataset
The Py-CoMSIA implementation recently demonstrated the effectiveness of PLS regression in 3D-QSAR by validating against the classic steroid benchmark dataset originally used in CoMSIA methodology development [19]. Using the standard steric, electrostatic, and hydrophobic (SEH) field combination with PLS regression, the implementation achieved a cross-validated q² of 0.609 with three optimal components, closely matching the original Sybyl implementation's q² of 0.665 with four components [19]. The conventional correlation coefficient r² reached 0.917, indicating excellent model fit, while the predictive r² of 0.40 for an external test set confirmed robust generalization capability [19]. When extending the analysis to include hydrogen bond donor and acceptor fields (SEHAD), the model maintained statistical significance with q² = 0.630 and r² = 0.898, though predictive performance slightly decreased (r²pred = 0.186), potentially due to descriptor overload or suboptimal alignment [19]. Field contribution analysis revealed the relative importance of different molecular properties, with electrostatic (0.534) and hydrophobic (0.316) fields dominating the SEH model, while all five fields contributed more balanced in the SEHAD model [19].
Table 1: Performance Metrics of PLS-Based CoMSIA Models on Benchmark Steroid Dataset
| Model Parameters | Published Sybyl (SEH) | Py-CoMSIA (SEH) | Py-CoMSIA (SEHAD) |
|---|---|---|---|
| q² (LOO-CV) | 0.665 | 0.609 | 0.630 |
| r² (conventional) | 0.937 | 0.917 | 0.898 |
| r²pred (test set) | 0.318 | 0.40 | 0.186 |
| Standard error (S) | 0.33 | 0.33 | 0.366 |
| Optimal components | 4 | 3 | 3 |
| Field contributions | |||
| Steric | 0.073 | 0.149 | 0.065 |
| Electrostatic | 0.513 | 0.534 | 0.258 |
| Hydrophobic | 0.415 | 0.316 | 0.154 |
| Hydrogen bond donor | - | - | 0.274 |
| Hydrogen bond acceptor | - | - | 0.248 |
α1A-Adrenergic Receptor Antagonists Study
In a comprehensive study on α1A-adrenergic receptor antagonists, researchers developed both CoMFA and CoMSIA models using pharmacophore-based molecular alignment and PLS regression [12]. The dataset comprised 44 compounds with binding affinities spanning four orders of magnitude (0.1-630 nM), divided into training (32 compounds) and test (12 compounds) sets [12]. Both models demonstrated exceptional predictive power, with identical leave-one-out cross-validated q² values of 0.840 [12]. The external predictive ability remained robust, with CoMFA achieving r²pred = 0.694 and CoMSIA reaching r²pred = 0.671 for the test set [12]. The CoMSIA approach incorporated five field types (steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor), with the resulting model highlighting the critical importance of electrostatic, hydrophobic, and hydrogen bonding interactions between ligands and the α1A-AR receptor [12]. The contour maps generated from these PLS-based models provided specific guidance for structural modifications to enhance antagonist activity, demonstrating the practical utility of this methodology in rational drug design [12].
Anticandida Pyrrole Derivatives Analysis
A CoMFA study on 3-aryl-4-[α-(1H-imidazol-1-yl)aryl methyl]pyrroles with anticandida activity further illustrates the application of PLS regression in 3D-QSAR [60]. The analysis utilized 33 compounds for model development and reserved 7 compounds for external validation [60]. The resulting PLS model demonstrated a strong fit with conventional r² = 0.964 and acceptable cross-validated predictive ability (q² = 0.598) [60]. The model identified key steric and electrostatic requirements for anticandida activity through coefficient contour maps, enabling researchers to rationalize the activity trends observed across the compound series [60]. This case study exemplifies how PLS regression can effectively handle 3D-QSAR data even with moderate-sized datasets, generating biologically meaningful models that guide the optimization of therapeutic agents against fungal pathogens [60].
Table 2: Comparative Performance of PLS-Based 3D-QSAR Models Across Various Applications
| Study | Method | Field Types | q² (LOO-CV) | r² | r²pred | Components |
|---|---|---|---|---|---|---|
| Steroids [19] | CoMSIA | SEH | 0.609 | 0.917 | 0.40 | 3 |
| Steroids [19] | CoMSIA | SEHAD | 0.630 | 0.898 | 0.186 | 3 |
| α1A-AR Antagonists [12] | CoMFA | SE | 0.840 | N/R | 0.694 | N/R |
| α1A-AR Antagonists [12] | CoMSIA | SEHDA | 0.840 | N/R | 0.671 | N/R |
| Anticandida Pyrroles [60] | CoMFA | SE | 0.598 | 0.964 | N/R | N/R |
Table 3: Essential Computational Tools for PLS-Based 3D-QSAR Modeling
| Resource Category | Specific Tools | Primary Function | Application in 3D-QSAR |
|---|---|---|---|
| Molecular Modeling | SYBYL [12], Schrödinger [19], MOE [19] | Comprehensive molecular modeling platforms | Historically standard for CoMFA/CoMSIA; provide integrated environments for alignment, field calculation, and PLS analysis |
| Open-Source Cheminformatics | RDKit [19], NumPy [19] | Open-source chemical informatics and numerical computing | Generate 3D structures, perform molecular alignment, and implement core CoMSIA algorithms |
| 3D-QSAR Specific | Py-CoMSIA [19] | Open-source Python implementation of CoMSIA | Provides accessible, flexible alternative to proprietary software for CoMSIA analysis |
| Visualization | PyVista [19] | 3D plotting and mesh analysis | Visualization of molecular structures, alignment, and CoMSIA similarity maps |
| Statistical Analysis | PLS implementations in SYBYL [12] [60] | Multivariate statistical analysis | Core PLS regression for correlating field variables with biological activity |
| Alignment Tools | GALAHAD [12] | Pharmacophore identification and molecular alignment | Generates pharmacophore models and aligns compounds for 3D-QSAR studies |
Troubleshooting and Technical Considerations
Addressing Common Implementation Challenges
Successful implementation of PLS regression in 3D-QSAR requires careful attention to several potential pitfalls. Molecular alignment represents the most critical and subjective step, with poor alignment consistently resulting in models with low predictive power [11]. When working with structurally diverse compounds, pharmacophore-based alignment methods often outperform common substructure approaches [12]. Descriptor selection and collinearity must be carefully managed, as including too many field types without sufficient observations can lead to overfitting despite the dimensional reduction capabilities of PLS [19]. The optimal number of components must be determined rigorously through cross-validation rather than arbitrary selection, as too few components underfit the data while too many capture noise rather than signal [60]. Statistical significance should be verified through multiple validation techniques including leave-one-out cross-validation, bootstrapping, and external test sets to ensure model robustness [12] [60]. Finally, model interpretation requires correlation of coefficient contour maps with structural features of known active compounds to ensure physicochemical plausibility [11].
Advanced Methodological Considerations
For researchers seeking to enhance their PLS-based 3D-QSAR models, several advanced methodological approaches merit consideration. Region focusing techniques can be employed to weight specific grid regions of greater potential importance, thereby improving signal-to-noise ratio in the field data [12]. SAMPLS algorithm implementation offers computational efficiency advantages for leave-one-out cross-validation with large descriptor sets [60]. Integration with other QSAR approaches such as conventional 2D-QSAR or machine learning methods can provide complementary insights, as demonstrated in studies on acylshikonin derivatives where principal component regression (PCR) outperformed PLS for specific descriptor sets [61]. Field energy cutoffs should be optimized rather than relying solely on default values, as excessively high cutoff values in CoMFA can eliminate potentially meaningful interaction data [12]. Finally, bootstrapping provides valuable estimates of confidence intervals for field contributions, helping to distinguish robust effects from statistical artifacts [60].
Three-dimensional Quantitative Structure-Activity Relationship (3D-QSAR) techniques, primarily Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), have become cornerstone methodologies in modern computer-aided drug design. These approaches quantitatively correlate the three-dimensional electronic and steric properties of molecules with their biological activities, providing interpretable models that guide the rational design of novel therapeutics. Unlike traditional 2D-QSAR, which relies on physicochemical descriptors, 3D-QSAR methods account for the spatial orientation of molecules, offering critical insights into the non-covalent interactions governing ligand-receptor binding. This article details practical protocols and presents concrete case studies demonstrating the successful application of CoMFA and CoMSIA across two critical target classes: HIV-1 protease in antiviral therapy and kinase inhibitors in oncology, framing these applications within a broader thesis on 3D-QSAR methodology.
Core Principles of CoMFA and CoMSIA
Theoretical Foundations
The fundamental premise of 3D-QSAR is that biological activity can be correlated with ligand-receptor interaction energies, which are mimicked by probing the molecular fields surrounding a set of aligned ligand structures.
- CoMFA (Comparative Molecular Field Analysis): This method characterizes molecular structures using steric (Lennard-Jones potential) and electrostatic (Coulombic potential) fields sampled by a probe atom at thousands of points in a 3D grid surrounding the aligned molecules [62]. The genius of CoMFA lies in using these interaction energies as descriptors for regression analysis, effectively mapping the forces a ligand would experience upon approaching its receptor.
- CoMSIA (Comparative Molecular Similarity Indices Analysis): An extension of CoMFA, CoMSIA employs a Gaussian function to calculate similarity indices, avoiding the singularities inherent in CoMFA's potential functions. Notably, CoMSIA can incorporate up to five distinct molecular fields: steric, electrostatic, hydrophobic, and hydrogen-bond donor and acceptor fields [16] [25] [63]. This provides a more nuanced and often more robust model of binding interactions.
The General 3D-QSAR Workflow
The application of these methods follows a systematic workflow, from data preparation to model deployment, as illustrated below.
Case Study 1: Application in HIV-1 Protease Inhibitor Design
Background and Rationale
HIV-1 protease is an essential enzyme for viral replication and maturation, making it a prime target for antiretroviral therapy [64]. The challenge of drug resistance necessitates the continuous design of new inhibitors. 3D-QSAR has been instrumental in understanding the structural requirements for effective inhibition.
Protocol: Building a Robust 3D-QSAR Model for Cyclic Urea Derivatives
Objective: To construct predictive CoMFA and CoMSIA models for a set of 120 cyclic urea-based HIV-1 protease inhibitors [65].
Software: SYBYL molecular modeling software.
Detailed Methodology:
Structure Preparation and Alignment:
- Sketch 3D structures of all 120 cyclic urea molecules.
- Perform energy minimization using the Tripos force field with a distance-dependent dielectric and the Powell conjugate gradient algorithm (convergence criterion of 0.001 kcal/(mol·Å)) [16].
- Calculate partial atomic charges using the Gasteiger-Hückel method.
- For alignment, use the field-fit method. The most active molecule is used as a template, and all others are superimposed by minimizing the RMSD of their steric and electrostatic fields relative to the template [16].
Field Calculation and PLS Analysis:
- Place the aligned molecules in a 3D grid with a spacing of 2.0 Å.
- For CoMFA, use an sp³ carbon probe with a +1 charge to calculate steric (Lennard-Jones potential) and electrostatic (Coulombic potential) fields. Set a cutoff value of 30 kcal/mol for both fields [16].
- For CoMSIA, calculate five similarity descriptor fields: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor, using a common probe atom and an attenuation factor of 0.3 [16] [64].
- Use the Partial Least Squares (PLS) algorithm to derive the correlation between the field descriptors and the experimental inhibitory activity (pIC50 or pKi). Apply a column filtering of 0.3 kcal/mol to reduce noise [16].
Model Validation:
- Evaluate the model's internal predictive ability using leave-one-out (LOO) cross-validation, yielding a cross-validated coefficient ( q^2 ) [65].
- Validate the model's external predictive power by using a test set of 60 molecules that were not included in the model building, yielding a predictive correlation ( r^2_{pred} ) [65].
Key Findings and Application
The study yielded highly predictive models. The CoMFA model achieved a non-cross-validated ( r^2 ) of 0.983 and a predictive ( r^2{pred} ) of 0.684 for the test set [65]. To rigorously test the model's generalizability, it was used to predict the activity of 25 non-cyclic urea inhibitors, achieving a remarkable ( r^2{pred} ) of 0.61 for CoMFA, demonstrating its utility beyond the chemical scaffold used for training [65].
Table 1: Statistical Results of 3D-QSAR Models for HIV-1 Protease Inhibitors
| Model | Number of Compounds | ( q^2 ) | ( r^2 ) | ( r^2_{pred} ) | Field Contributions |
|---|---|---|---|---|---|
| CoMFA (Cyclic Ureas) [65] | 120 (60 training) | 0.598 | 0.983 | 0.684 | Steric, Electrostatic |
| CoMSIA (Cyclic Ureas) [65] | 120 (60 training) | 0.674 | 0.985 | 0.640 | Steric, Electrostatic, Hydrophobic, H-Bond Donor/Acceptor |
| CoMSIA (Cyclic Ureas) [64] | 34 (27 training) | 0.586 | 0.931 | 0.973 | Steric (21.6%), Electrostatic (31.0%), Hydrophobic (25.8%), H-Bond Donor (13.1%), H-Bond Acceptor (8.5%) |
The contour maps provided clear design guidance. For example, a CoMSIA study on 34 cyclic ureas revealed that hydrogen-bond donor and acceptor groups near the carbonyl oxygen of the cyclic urea core were crucial for interacting with the ASP25 residues in the protease active site, a finding confirmed by molecular docking [64]. This information has been used to design new analogs with optimized binding interactions, some of which show predicted activities higher than the parent compounds [64] [66].
Case Study 2: Application in Kinase Inhibitor Design (Bcr-Abl)
Background and Rationale
The Bcr-Abl fusion protein is a constitutively active tyrosine kinase that drives Chronic Myeloid Leukemia (CML). While inhibitors like imatinib are effective, resistance—particularly from the T315I "gatekeeper" mutation—remains a major clinical challenge [10]. This case study explores the use of 3D-QSAR to design novel purine-based Bcr-Abl inhibitors.
Protocol: Fragment-Based 3D-QSAR for Purine Derivatives
Objective: To perform CoMFA, CoMSIA, and Topomer CoMFA on a database of 58 purine derivatives to guide the design of new Bcr-Abl inhibitors [10].
Software: SYBYL-X 2.0.
Detailed Methodology:
Data Set and Molecular Modeling:
- A dataset of 58 compounds with known Bcr-Abl half-maximal inhibitory concentration (IC50) values was compiled. The biological activity was converted to pIC50 (-logIC50) for QSAR analysis.
- The 3D structures of all inhibitors were sketched and energy-minimized using the Tripos force field.
Alignment and Field Calculation:
- For Topomer CoMFA, each molecule was automatically fragmented into a common skeleton (e.g., the purine core) and two variable regions (R1 and R2). This automated alignment enhances reproducibility and computational efficiency [67].
- Standard settings were used: a 3D grid with 2.0 Å spacing and an sp³ carbon probe with a +1 charge to calculate steric and electrostatic fields.
Model Construction and Validation:
- PLS regression was used to build the QSAR models.
- The models were rigorously validated using both internal cross-validation (( q^2 )) and external test set prediction (( r^2_{pred} )).
Key Findings and Application
The generated CoMFA and CoMSIA models exhibited excellent predictive power. The optimal CoMSIA model, which incorporated steric, electrostatic, hydrophobic, hydrogen-bond donor, and hydrogen-bond acceptor fields, yielded a ( q^2 ) of 0.734 and a high ( r^2_{pred} ) of 0.891 [10].
Table 2: Statistical Results of 3D-QSAR Models for Bcr-Abl Kinase Inhibitors
| Model | ( q^2 ) | ( r^2 ) | ( r^2_{pred} ) | Field Contributions |
|---|---|---|---|---|
| CoMFA [10] | 0.679 | 0.983 | 0.884 | Steric (46.3%), Electrostatic (53.7%) |
| CoMSIA (S+E+H+D+A) [10] | 0.734 | 0.985 | 0.891 | Steric (12.3%), Electrostatic (41.4%), Hydrophobic (27.6%), H-Bond Donor (11.2%), H-Bond Acceptor (7.5%) |
The contour maps provided critical structural insights. A large green steric favorable contour near the C4-position of the left-wing phenyl ring indicated that bulky substituents in this region enhance activity, likely by improving van der Waals contacts with a hydrophobic pocket in the kinase [10] [63]. This guided the design of novel purine derivatives. Subsequent synthesis and biological testing confirmed the predictions, with several new compounds (e.g., 7a and 7c) demonstrating IC50 values superior to imatinib. Notably, some designed compounds (7e and 7f) also showed significant activity against the resistant T315I mutant, highlighting the power of this approach in addressing drug resistance [10].
Integrated Protocol: A Standardized Workflow for 3D-QSAR Analysis
The following diagram and text summarize a consolidated, best-practice protocol derived from the case studies.
Phase 1: Preparation
- Compound Selection: Curate a data set of 20-100+ compounds with consistent biological activity data. Divide into a training set (for model building) and a test set (for validation) [62].
- 3D Structure Building: Sketch 3D structures and perform geometry optimization via energy minimization (e.g., Tripos force field, Powell conjugate gradient algorithm, convergence = 0.001 kcal/(mol·Å)) [16].
- Molecular Alignment: This is the most critical step. Align molecules based on a common substructure or pharmacophore. The field-fit method, which minimizes the RMSD of molecular fields to a template, is highly effective [16].
Phase 2: Modeling & Validation
- Descriptor Calculation: Place aligned molecules in a 2.0 Å grid. Calculate CoMFA (steric, electrostatic) and/or CoMSIA (up to five fields) descriptors using standard probe atoms [16] [67].
- PLS Regression: Use PLS to correlate the thousands of field descriptors with biological activity. Apply column filtering (e.g., 0.3-2.0 kcal/mol) to reduce noise [16].
- Model Validation: Internally validate with LOO cross-validation (( q^2 > 0.5 ) is acceptable; >0.7 is excellent). Externally validate by predicting the test set (( r^2_{pred} > 0.5 ) indicates good predictive ability) [65] [10] [63].
Phase 3: Application
- Contour Map Analysis: Visualize the results as 3D contour maps surrounding the aligned molecules. These maps highlight regions where specific molecular properties (e.g., bulky groups, hydrogen bond donors) increase or decrease activity.
- Rational Design & Prediction: Use the contour maps to propose new chemical structures with optimized properties. The validated model can then predict their activities prior to costly synthesis and testing [10] [64].
The Scientist's Toolkit: Essential Research Reagents & Software
Table 3: Key Resources for 3D-QSAR Studies
| Category | Item / Software | Specific Function in 3D-QSAR |
|---|---|---|
| Commercial Software Suites | SYBYL (Tripos, Inc.) [16] [10] | Integrated environment for molecular modeling, alignment, and performing CoMFA/CoMSIA analyses. |
| Docking & Simulation | AutoDock [64] | Molecular docking to elucidate binding modes and conformations for alignment or model interpretation. |
| Force Fields | Tripos Force Field [16] | Energy minimization and conformational analysis of ligand structures prior to alignment. |
| Charge Calculation Methods | Gasteiger-Hückel [16], Gasteiger-Marsili [68], AM1-ESP [68] | Calculation of partial atomic charges, which critically influence electrostatic field calculations. |
| Probe Atoms | sp³ Carbon atom (+1 charge) [16] [67] | Standard probe for calculating steric and electrostatic interaction fields in CoMFA. |
| Algorithmic Tools | Partial Least Squares (PLS) [16] [62] | Core regression method for handling the high number of field descriptors relative to compounds. |
| Validation Metrics | Leave-One-Out (LOO) ( q^2 ), Predictive ( r^2_{pred} ) [65] [10] | Quantitative metrics for assessing the internal and external predictive power of the 3D-QSAR model. |
The detailed case studies of HIV-1 protease and Bcr-Abl kinase inhibitors underscore the profound practical impact of CoMFA and CoMSIA in accelerating drug discovery. These 3D-QSAR techniques successfully transcend chemical scaffolds, as evidenced by models trained on cyclic ureas accurately predicting activities of structurally distinct inhibitors, and provide a quantifiable, visual blueprint for molecular design. The provided integrated protocol offers a standardized framework that researchers can adapt to their specific targets. The true power of these methods is fully realized when they are integrated with complementary computational techniques—such as molecular docking to inform alignment, dynamics simulations to account for protein flexibility, and ADMET prediction to optimize pharmacokinetics—and, most importantly, when they are iteratively refined with experimental feedback. This synergy between in silico modeling and wet-lab experimentation continues to make 3D-QSAR an indispensable tool in the ongoing quest to develop novel, effective therapeutics for complex diseases.
Overcoming Common Challenges in 3D-QSAR Studies
Addressing Alignment Sensitivity in CoMFA Models
Comparative Molecular Field Analysis (CoMFA) is a foundational technique in three-dimensional quantitative structure-activity relationship (3D-QSAR) modeling that correlates the steric and electrostatic fields of molecules with their biological activities [11]. A critical methodological vulnerability of traditional CoMFA is its high sensitivity to molecular alignment, where small changes in the spatial orientation of molecules can significantly impact model performance and predictive accuracy [69]. This protocol examines the sources of alignment sensitivity and provides detailed methodologies for addressing this challenge through improved alignment strategies and alternative approaches.
The fundamental issue stems from CoMFA's reliance on calculating interaction energies using a probe atom at regularly spaced grid points surrounding aligned molecules. Unlike later methods such as Comparative Molecular Similarity Indices Analysis (CoMSIA), which uses Gaussian functions to create more continuous fields, CoMFA employs discrete energy cutoffs that can create abrupt field changes with minor positional adjustments [69]. This technical implementation makes the quality of molecular superposition a primary determinant of model robustness.
Understanding the Roots of Alignment Sensitivity
Technical Foundations of Sensitivity
CoMFA models calculate steric (Lennard-Jones) and electrostatic (Coulombic) fields using a probe atom on a 3D grid surrounding aligned molecules [11]. The discrete nature of these calculations, combined with energy truncation limits (typically ±30 kcal/mol), creates inherent sensitivity to molecular positioning [69]. When molecules are misaligned, the same structural features may map to different grid points, introducing noise that obscures genuine structure-activity relationships.
The CoMSIA method, developed as an advancement to CoMFA, addresses this limitation by employing a Gaussian-type function for field calculations instead of the traditional Coulomb and Lennard-Jones potentials [69]. This fundamental difference in approach makes CoMSIA "less sensitive to factors that traditionally complicated CoMFA, such as molecular alignment, grid spacing, and probe atom selection" [69]. The Gaussian function ensures that small conformational differences produce proportionally small changes in similarity indices, creating more continuous and alignment-tolerant field distributions.
Practical Consequences of Misalignment
In practical applications, alignment inconsistencies can severely compromise model predictivity. Poor alignment introduces artificial variance in descriptor values that does not correspond to actual biological activity differences. This noise manifests as reduced Q² values in cross-validation and poor external prediction accuracy on test compounds. The 3D-SDAR technique, an alignment-independent alternative, demonstrated that avoiding complex alignment procedures could achieve predictive performance comparable to alignment-dependent methods while requiring only 3-7% of the computational time [48].
Comparative Analysis of Alignment Methods
Table 1: Comparison of Molecular Alignment Strategies for CoMFA
| Method | Key Principle | Advantages | Limitations | Best Applications |
|---|---|---|---|---|
| Common Scaffold (MCS) | Superposition based on largest shared structural framework [11] [70] | Intuitive; preserves pharmacophore geometry; reproducible | Limited to compounds with significant structural similarity | Congeneric series with clear core structure |
| Pharmacophore-Guided | Alignment based on 3D arrangement of key functional groups [70] | Biologically relevant; can handle diverse chemotypes | Requires prior knowledge of binding pharmacophore | Diverse sets with known key interactions |
| Shape-Based/Overlay | Maximization of molecular volume overlap [70] | No need for common substructure; reflects binding site constraints | May emphasize irrelevant steric bulk | Targets with well-defined binding pockets |
| Template-Based | Alignment to reference molecule(s) [48] | Straightforward; uses known active conformations | Reference selection critical; may bias model | Datasets with well-characterized lead compounds |
| 2D-to-3D Direct | Non-aligned conformations from 2D structure conversion [48] | Fast; avoids alignment subjectivity; high throughput | May not reflect bioactive conformation | Large diverse datasets; initial screening models |
Experimental Protocols for Robust Alignment
Common Scaffold Alignment Protocol
Objective: Achieve consistent molecular superposition using maximum common substructure (MCS) to minimize alignment-related variance in CoMFA models.
Materials and Software:
- Molecular dataset with associated biological activities
- SYBYL-X 2.1.1 or equivalent molecular modeling software
- RDKit (Open-source cheminformatics toolkit)
Procedure:
- Identify Maximum Common Substructure: Using RDKit's MCS algorithm, identify the largest common substructure shared across all molecules in the dataset [11].
- Conformer Generation: Generate 3D conformations for each molecule using RDKit's ETKDG method or SYBYL's Concord module.
- Geometry Optimization: Optimize molecular geometries using molecular mechanics force fields (MMFF94 or Tripos) with gradient convergence criterion of 0.05 kcal/mol·Å.
- Structural Alignment: Superimpose molecules by fitting the identified MCS atomic coordinates to a reference framework molecule using least-squares fitting.
- Alignment Quality Assessment: Calculate root-mean-square deviation (RMSD) values for the aligned MCS atoms. Accept alignments with RMSD < 0.5 Å for the core structure [70].
- CoMFA Field Calculation: Proceed with standard CoMFA analysis using 2.0 Å grid spacing and sp³ carbon probe with +1 charge.
Troubleshooting Notes: If MCS is too small (<5 heavy atoms), consider using pharmacophore-guided alignment instead. For flexible molecules, apply conformational searching to identify low-energy conformers that permit reasonable overlay.
Pharmacophore-Guided Alignment Protocol
Objective: Align molecules based on 3D pharmacophoric features to reflect biologically relevant interactions while minimizing alignment arbitrariness.
Materials and Software:
- Molecular dataset with known or hypothesized pharmacophore
- Molecular operating environment (MOE) or Schrodinger Maestro
- Structure-based pharmacophore generation tools
Procedure:
- Pharmacophore Hypothesis Generation: Derive pharmacophore features from:
- Protein-ligand crystal structure (if available)
- Common features among known active compounds
- Receptor surface analysis
- Feature Definition: Specify 3-5 key pharmacophore elements (hydrogen bond donors/acceptors, hydrophobic regions, charged groups, aromatic rings).
- Conformer Sampling: Generate multiple low-energy conformers for each compound (maximum 50 conformers per molecule, energy window 10 kcal/mol).
- Pharmacophore Matching: Align compounds by fitting their pharmacophoric features to the hypothesis using constrained optimization.
- Consensus Scoring: Rank alignments using combination of pharmacophore fit score and strain energy.
- Validation: Verify alignment by examining positional consistency of key functional groups known to be critical for activity.
Quality Control: The resulting alignment should place pharmacophore elements within 1.0 Å of their hypothesized positions. Cross-validate with known structure-activity relationships.
Diagram 1: Molecular alignment and CoMFA workflow. The process involves parallel alignment strategies with quality checkpoints to ensure robust model development.
Alignment-Independent Alternatives
CoMSIA Implementation Protocol
CoMSIA represents the most direct alternative to address CoMFA's alignment limitations while maintaining the 3D-QSAR paradigm [69]. By replacing the traditional potential functions with Gaussian-type similarity indices, CoMSIA creates smoother field distributions that are less susceptible to alignment variations.
Implementation Steps:
- Molecular Preparation: Follow the same conformational generation and initial alignment procedures as CoMFA.
- Field Selection: Choose from five available field types: steric (S), electrostatic (E), hydrophobic (H), hydrogen bond donor (D), and hydrogen bond acceptor (A).
- Parameter Settings: Use default attenuation factor (0.3) and 1.0 Å grid spacing for initial models.
- Model Construction: Apply partial least squares (PLS) regression with the same validation protocols as CoMFA.
Recent Implementation: The open-source Py-CoMSIA package provides an accessible implementation in Python, using RDKit for calculations and PyVista for visualization [69]. This implementation successfully replicated classical CoMSIA results on benchmark steroid datasets, demonstrating its viability as a CoMFA alternative.
3D-SDAR and Other Alignment-Free Approaches
The 3D-Spectral Data-Activity Relationship (3D-SDAR) technique offers a fundamentally different approach that completely bypasses molecular alignment [48]. This method represents each compound by a unique "fingerprint" constructed from carbon atom pairs, with dimensions based on their NMR chemical shifts and interatomic distances.
Key Advantage: A study comparing 3D-SDAR performance using different conformational strategies found that "the best model using 2D>3D (imported directly from ChemSpider) produced R²Test = 0.61," which was superior to energy-minimized and conformation-aligned models while requiring only 3-7% of the computational time [48].
Validation and Quality Control Measures
Alignment Quality Assessment
Quantitative Metrics:
- RMSD of Core Atoms: Calculate root-mean-square deviation of heavy atoms in the common scaffold (<0.5 Å acceptable).
- Pharmacophore Feature Overlap: Measure distance between corresponding pharmacophore elements (<1.0 Å acceptable).
- Volume Overlap: Compute intersection volume of molecular van der Waals surfaces (>70% overlap desirable).
Visual Inspection:
- Examine aligned structures from multiple viewpoints to identify obvious misalignments.
- Verify conservation of known pharmacophoric features across the series.
- Check that key functional groups maintain consistent spatial relationships.
Model Robustness Validation
Statistical Validation Protocols:
- Cross-Validation: Leave-one-out (LOO) or leave-multiple-out (LMO) with Q² > 0.5 considered acceptable.
- External Validation: Reserve 20-30% of compounds as external test set with R²pred > 0.6.
- Y-Randomization: Randomize activity values to ensure model not correlating noise (should yield low Q²).
- Applicability Domain: Define chemical space coverage using Williams plots and leverage statistics.
Table 2: Research Reagent Solutions for Alignment-Sensitive CoMFA Studies
| Tool/Category | Specific Software/Packages | Primary Function | Alignment Relevance |
|---|---|---|---|
| Commercial Molecular Modeling | SYBYL-X [71], Schrodinger Maestro, MOE | Comprehensive molecular modeling platforms | Built-in alignment tools; automated CoMFA/CoMSIA workflows |
| Open-Cheminformatics | RDKit [69], Open3DALIGN | Open-source chemical analysis | MCS identification; conformer generation; scripting flexibility |
| Specialized 3D-QSAR | Py-CoMSIA [69] | Python-based CoMSIA implementation | Alignment-tolerant alternative to CoMFA; open-source accessibility |
| Alignment Algorithms | Phase Shape Alignment, ROCS | Pharmacophore and shape-based superposition | Advanced alignment methods beyond simple atom fitting |
| Validation Tools | Cross-validation utilities, y-randomization scripts | Model robustness assessment | Quantifying alignment impact on model stability |
Addressing alignment sensitivity is crucial for developing robust CoMFA models with reliable predictive power. Based on the methodologies examined, the following implementation pathway is recommended:
For congeneric series with clear common scaffold, employ MCS-based alignment with rigorous RMSD quality control. For structurally diverse compounds with hypothesized pharmacophore, use pharmacophore-guided alignment. When facing significant alignment challenges or computational constraints, implement CoMSIA or 3D-SDAR as alignment-tolerant alternatives.
The integration of multiple alignment strategies with comprehensive validation provides the most robust approach to managing alignment sensitivity in CoMFA studies. The scientific literature demonstrates that acknowledging and systematically addressing this methodological vulnerability leads to more reliable 3D-QSAR models that effectively guide drug discovery efforts.
Optimizing CoMSIA Parameters for Diverse Chemical Datasets
Within the broader context of developing robust Three-Dimensional Quantitative Structure-Activity Relationship (3D-QSAR) protocols, Comparative Molecular Similarity Indices Analysis (CoMSIA) stands as a pivotal methodology for understanding the intricate relationships between molecular structure and biological activity [19] [14]. Unlike its predecessor, Comparative Molecular Field Analysis (CoMFA), CoMSIA incorporates a broader spectrum of molecular interaction fields and employs a Gaussian function for descriptor calculation, thereby avoiding abrupt potential cutoffs and enhancing model interpretability [14] [69]. The core strength of CoMSIA lies in its ability to map five distinct molecular fields—steric, electrostatic, hydrophobic, and hydrogen bond donor and acceptor—onto a common grid, providing a holistic view of the interaction landscape [19]. However, the predictive power and robustness of CoMSIA models are profoundly influenced by the careful optimization of its underlying parameters, a process critical for adapting the methodology to chemically diverse datasets in modern drug discovery programs [72]. This document outlines detailed protocols and application notes for the systematic optimization of CoMSIA parameters, ensuring the development of reliable, predictive models that can effectively guide lead optimization.
Core CoMSIA Parameters and Optimization Strategies
The performance of a CoMSIA model is governed by several interlinked parameters. Optimizing these parameters is essential for creating a model that is both statistically sound and chemically meaningful. The key parameters and strategies for their optimization are summarized in the table below.
Table 1: Key CoMSIA Parameters and Optimization Strategies
| Parameter | Description | Default Value(s) | Optimization Strategy |
|---|---|---|---|
| Molecular Fields | Physicochemical properties evaluated (Steric, Electrostatic, Hydrophobic, H-Bond Donor, H-Bond Acceptor) | Steric, Electrostatic | Systematically test different field combinations (e.g., SE, SEH, SEHAD); select based on cross-validated ( q^2 ) and field contribution interpretability [19] [73]. |
| Attenuation Factor (α) | Defines the width of the Gaussian function, controlling the rate of decay of the similarity index with distance. | 0.3 | Evaluate a range of values (e.g., 0.1, 0.2, 0.3, 0.4); higher values create steeper, more localized fields [19] [73]. |
| Grid Spacing | The resolution of the 3D lattice surrounding the aligned molecules. | 2.0 Å | Test smaller spacings (e.g., 1.0 Å, 1.5 Å) for finer detail, balancing computational cost and potential for overfitting [19]. |
| Region Focusing | A technique to emphasize descriptor regions with high information content. | Not applied | Use methods like GOLPE or ( q^2 )-guided region selection to identify and weight critical regions, improving signal-to-noise [14]. |
| Statistical Method | The algorithm used to correlate field descriptors with biological activity. | Partial Least Squares (PLS) | For complex, non-linear relationships, integrate Machine Learning algorithms (e.g., Gradient Boosting, SVM) with hyperparameter tuning [72] [5]. |
Advanced Optimization via Machine Learning Integration
Traditional CoMSIA relies on Partial Least Squares (PLS) regression, which can be limited when handling the thousands of descriptors generated and their potential non-linear relationships with activity [72]. Integrating machine learning (ML) provides a powerful avenue for optimization.
A robust ML-based CoMSIA protocol involves:
- Feature Selection: Apply techniques like Recursive Feature Elimination (RFE) or SelectFromModel to reduce descriptor dimensionality and minimize noise [72] [74].
- Algorithm Selection: Test non-linear estimators such as Gradient Boosting Regression (GBR), Support Vector Machines (SVM), or Random Forest (RF) to capture complex structure-activity relationships [72] [5].
- Hyperparameter Tuning: Use methods like GridSearchCV to systematically optimize the ML algorithm's parameters (e.g.,
learning_rate,max_depthfor GBR) [72].
For instance, a study on antioxidant peptides demonstrated that a GBR model coupled with GB-RFE feature selection (with tuned hyperparameters: learning_rate=0.01, max_depth=2, n_estimators=500) significantly outperformed the traditional PLS model, yielding a superior ( R^2_{test} ) of 0.759 compared to 0.575 [72]. This highlights the potential of ML to mitigate overfitting and enhance predictive performance.
Experimental Protocol for CoMSIA Model Development and Validation
This section provides a detailed, step-by-step protocol for building and validating a optimized CoMSIA model, from data preparation to final application.
Dataset Curation and Molecular Preparation
- Data Collection: Compile a dataset of chemical structures and their corresponding biological activities (e.g., IC₅₀, Ki) from reliable sources. Ensure activity data is in a consistent unit and scale (e.g., pKi = -logKi) [72] [75].
- Structure Optimization: Generate high-quality 3D molecular structures using tools like RDKit or commercial software. Conduct a conformational search to identify the putative bioactive conformation for each molecule. Common methods include systematic search or distance-comparison techniques [72].
- Molecular Alignment: This is a critical step for all alignment-dependent 3D-QSAR methods. Align molecules based on a common scaffold, pharmacophore hypothesis, or molecular docking poses. The choice of alignment rule directly impacts the model's validity and must be carefully considered [19] [14].
CoMSIA Field Calculation and Model Building
- Grid Generation: Construct a 3D lattice that encompasses all aligned molecules. A padding of 4 Å beyond the molecular dimensions is typically sufficient. The grid spacing should be set according to the optimization studies (e.g., 1.0 or 2.0 Å) [19] [69].
- Descriptor Calculation: Using a chosen set of molecular fields and an attenuation factor (default 0.3), calculate the CoMSIA similarity indices at each grid point for every molecule in the dataset [19] [14].
- Data Splitting: Divide the dataset into a training set (≈80%) for model development and a test set (≈20%) for external validation. The Kennard-Stone algorithm can be used to ensure representative splitting of the chemical space [75].
Model Validation and Interpretation
- Internal Validation: Use the training set to build a model and assess its internal predictive ability via Leave-One-Out (LOO) cross-validation. The cross-validated correlation coefficient ( q^2 ) is a key metric, with a value > 0.5 generally considered acceptable [19] [76]. ( q^2 = 1 - \frac{\sum{(Y{pred} - Y{actual})^2}}{\sum{(Y{actual} - \bar{Y}{actual})^2}} )
- External Validation: Predict the activity of the external test set molecules that were excluded from model building. The predictive ( r^2 ) (( r^2_{pred} )) should be calculated to evaluate the model's true generalizability [19] [75].
- Contour Map Analysis: Visualize the CoMSIA field coefficients as 3D contour maps. These maps highlight regions in space where specific molecular properties (e.g., steric bulk, hydrogen bond donors) are favorably or unfavorably correlated with biological activity, providing critical insights for molecular design [19] [14].
The following workflow diagram illustrates the complete CoMSIA model development process.
The Scientist's Toolkit: Essential Research Reagents and Software
Successful implementation of the CoMSIA protocol requires a suite of software tools and computational resources. The table below catalogues key solutions available to researchers.
Table 2: Essential Research Reagent Solutions for CoMSIA Studies
| Tool Name | Type | Key Function(s) | License/Status |
|---|---|---|---|
| Py-CoMSIA [19] [69] | Python Library | Open-source implementation of CoMSIA; calculates similarity indices, performs PLS analysis, and enables visualization. | Open-Source |
| RDKit [19] [75] | Cheminformatics Library | Handles molecular I/O, 3D structure generation, conformational analysis, and descriptor calculation. | Open-Source |
| Sybyl [19] [73] | Molecular Modeling Suite | The classic commercial platform for CoMFA/CoMSIA, providing integrated tools for alignment, field calculation, and statistical analysis. | Commercial |
| Schrödinger Suite [19] | Molecular Modeling Suite | Modern commercial platform that includes robust implementations of 3D-QSAR methods like CoMSIA within a comprehensive drug discovery environment. | Commercial |
| Scikit-learn [72] | Python ML Library | Provides a wide array of feature selection methods (RFE) and machine learning algorithms (GBR, SVM, RF) for building non-linear QSAR models. | Open-Source |
| Open Molecules 2025 (OMol25) [77] | Reference Dataset | A massive dataset of molecular simulations for training machine learning interatomic potentials, useful for advanced method development. | Open Access |
Application Notes and Case Studies
Case Study: Benchmarking with a Steroid Dataset
The open-source Py-CoMSIA library was validated using the classic steroid benchmark dataset. The model, built with steric, electrostatic, and hydrophobic (SEH) fields, a grid spacing of 1 Å, padding of 4 Å, and an attenuation factor of 0.3, yielded a ( q^2 ) of 0.609 and a predictive ( r^2 ) of 0.40. These results were comparable to the original Sybyl-based analysis (( q^2 ) = 0.665, predictive ( r^2 ) = 0.318), demonstrating the validity of the open-source implementation and the robustness of the standard parameter set [19] [69]. This case underscores the importance of using well-characterized benchmark sets to calibrate new tools and protocols.
Application Note: Handling Peptide Datasets with ML-CoMSIA
A study on lipid antioxidant tripeptides (FTC dataset) exemplifies the need for advanced optimization. The traditional PLS-based CoMSIA model showed suboptimal predictive power (( R^2{test} = 0.575 )). By integrating machine learning—specifically, Gradient Boosting Regression with Recursive Feature Elimination and hyperparameter tuning—the researchers achieved a superior model (( R^2{test} = 0.759 )). This ML-driven model was successfully used to screen and identify novel antioxidant peptides from the Tryptophyllin L family, which were subsequently synthesized and experimentally validated [72]. This application note highlights that for complex or noisy datasets, moving beyond default PLS to an ML-optimized workflow can be critical for generating useful predictive models.
Selecting Appropriate Probe Atoms and Grid Parameters
In the realm of three-dimensional quantitative structure-activity relationship (3D-QSAR) studies, particularly in Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), the selection of probe atoms and grid parameters is a foundational step that directly influences the predictive accuracy and interpretability of the models [11]. These technical choices govern how molecular interaction fields (MIFs) are sampled around aligned molecules, effectively translating 3D molecular structures into quantitative descriptors for statistical analysis [78]. Within the broader context of 3D-QSAR protocol development, optimal parameter selection ensures that the calculated fields accurately capture the essential physicochemical properties relevant to biological activity, while minimizing computational artifacts and noise [19] [79]. This protocol details the systematic selection and application of these critical parameters to empower researchers in constructing robust and predictive models for drug discovery.
Theoretical Background and Key Concepts
The Role of Probes and Grids in 3D-QSAR
In grid-based 3D-QSAR methods, the molecular environment is probed within a defined lattice that encloses the aligned molecules [11]. A probe atom or group is placed at each point in this 3D grid, and its hypothetical interactions with every atom of each molecule are calculated [4]. This process generates a set of field values for each molecule, which constitute the independent variables in the QSAR model [78]. The grid parameters—including spacing, extent, and placement—control the resolution and coverage of this molecular sampling. The choice of probe defines the specific physicochemical property being mapped, such as steric bulk or electrostatic potential [79]. Therefore, the interplay between probe and grid determines the fidelity of the molecular representation and the subsequent biological insights that can be derived from the contour maps [11].
Comparison of CoMFA and CoMSIA Approaches
While both CoMFA and CoMSIA rely on probes and grids, their fundamental calculation methods differ, leading to distinct practical considerations:
CoMFA (Comparative Molecular Field Analysis) uses a Lennard-Jones potential for steric fields and a Coulombic potential for electrostatic fields [4] [80]. This approach can lead to abrupt, discontinuous field changes and is highly sensitive to molecular alignment and grid positioning [19] [78].
CoMSIA (Comparative Molecular Similarity Indices Analysis) introduces a Gaussian-type function to calculate similarity indices [19]. This function produces continuous fields that are less sensitive to minor misalignments and grid shifts [19] [11]. CoMSIA also extends the analysis beyond steric and electrostatic fields to include hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields, providing a more holistic view of interactions [19] [79].
Table 1: Fundamental Differences Between CoMFA and CoMSIA Field Calculations
| Feature | CoMFA | CoMSIA |
|---|---|---|
| Field Calculation Method | Lennard-Jones & Coulombic potentials [4] | Gaussian-type similarity function [19] |
| Sensitivity to Alignment | High sensitivity [11] | More robust to small changes [11] |
| Field Types | Steric, Electrostatic [4] | Steric, Electrostatic, Hydrophobic, H-Bond Donor, H-Bond Acceptor [19] |
| Field Behavior | Discontinuous changes near van der Waals surfaces [19] | Smooth, continuous fields [19] |
Research Reagent Solutions: Computational Tools for 3D-QSAR
The following table catalogs essential software and computational tools used in modern 3D-QSAR studies for probe and grid setup, field calculation, and model analysis.
Table 2: Essential Software Tools for 3D-QSAR Studies
| Tool Name | Type/Category | Primary Function in Probe/Grid Setup |
|---|---|---|
| Sybyl (Tripos) | Commercial Software Suite | Classical platform for CoMFA/CoMSIA; provides tools for grid creation, field calculation, and statistical analysis [19]. |
| Open3DQSAR | Open-Source Software | Used for developing docking-based 3D-QSAR models; generates molecular interaction fields within a user-defined grid-box [80]. |
| Py-CoMSIA | Open-Source Python Library | A functional open-source implementation of CoMSIA, enabling grid-based field calculations and visualizations [19]. |
| RDKit | Open-Source Cheminformatics | Used for generating 3D molecular structures from 2D representations and for molecular geometry optimization [11]. |
| Glide (Schrödinger) | Molecular Docking Software | Used in structure-based alignment for 3D-QSAR; can provide bioactive conformations for grid placement [79] [81]. |
| AutoDock | Molecular Docking Software | Used to extract bioactive conformations from docking complexes for subsequent 3D-QSAR analysis [80]. |
Protocol: Probe Atom Selection
Standard Probe Specifications
The probe atom is defined by its atom type, charge, and other physicochemical properties, which determine the nature of the interaction field calculated [4]. The following standard probes are recommended for initial studies:
Table 3: Standard Probe Atoms and Properties for CoMFA and CoMSIA
| Field Type | Recommended Probe | Charge | Additional Properties | Application Note |
|---|---|---|---|---|
| CoMFA Steric | sp³ Carbon | 0 | Van der Waals radius ~1.52 Å | Measures steric hindrance using Lennard-Jones potential [4]. |
| CoMFA Electrostatic | Volumeless Probe | +1.0 | N/A | Measures Coulombic potential energy [80]. |
| CoMSIA Steric | sp³ Carbon | 0 | Atom type C.3 | Calculates similarity using a Gaussian function [19]. |
| CoMSIA Electrostatic | sp³ Carbon | +1.0 | Atom type C.3 | Calculates similarity using a Gaussian function [4]. |
| CoMSIA Hydrophobic | sp³ Carbon | 0 | Hydrophobicity +1.0 | Models affinity for lipophilic regions [4] [79]. |
| CoMSIA H-Bond Donor | sp³ Carbon | 0 | H-Bond Donor +1.0 | Identifies regions favorable for accepting a H-bond from the ligand [4] [79]. |
| CoMSIA H-Bond Acceptor | sp³ Carbon | 0 | H-Bond Acceptor +1.0 | Identifies regions favorable for donating a H-bond to the ligand [4] [79]. |
Practical Application Notes
- Probe Radius: A probe atom with a radius of 1.0 Å is typically used for CoMSIA field calculations [79].
- Charge Models: The choice of partial charge calculation method for the ligand atoms (e.g., Gasteiger-Huckel, MMFF94) can influence the electrostatic field. Studies have shown that the MMFF94 charge model can yield highly predictive CoMFA and CoMSIA models [79].
- Attenuation Factor: In CoMSIA, an attenuation factor (α) of 0.3 is commonly used as a default value in the Gaussian function for calculating distance-dependent similarities, providing a good balance between field resolution and smoothness [19] [79].
Protocol: Grid Parameter Configuration
Defining the Grid Lattice
The grid should encompass all aligned molecules with sufficient margin to sample relevant interaction regions.
- Grid Spacing: A spacing of 2.0 Å is widely used and provides a reasonable balance between computational cost and model resolution [4] [79]. A finer grid of 1.0 Å can be used for higher resolution but significantly increases the number of variables [19].
- Grid Region: The grid box should extend 4.0 Å beyond the combined molecular dimensions of all aligned molecules in every direction (x, y, z) [19]. This padding ensures complete coverage of the molecular ensemble.
- Grid Placement: The grid can be aligned based on the molecular aggregate or a common scaffold. For structure-based 3D-QSAR, the grid is often defined to enclose the bioactive conformations of ligands extracted from docking complexes [80] [79].
Energy Cutoffs and Field Treatment
To ensure numerical stability and eliminate irrelevant variables, apply energy cutoffs.
- Steric Cutoff: A fixed energy cutoff value of 30 kcal/mol is often applied to steric fields to avoid infinite values when the probe is very close to the molecule [79].
- Energy Thresholding: Molecular interaction field (MIF) energy values below a certain threshold (e.g., 0.05 kcal/mol) can be set to zero to reduce noise [80].
- Region Focusing: Advanced techniques like CoMFA Region Focusing (CoMFA-RF) can be used to weight specific lattice points to enhance the contribution of predictive regions and attenuate noisy variables [78].
The following diagram illustrates the logical workflow for setting up the grid and calculating molecular fields, integrating the key parameters discussed.
Experimental Validation and Benchmarking
Statistical Validation Metrics
After model building using Partial Least Squares (PLS) regression, rigorous validation is essential to ensure the model is both predictive and robust [4] [79]. Key metrics include:
- q² (q²): The cross-validated correlation coefficient (typically using Leave-One-Out method). A value > 0.5 is generally considered statistically significant [4] [79].
- r² (r²): The non-cross-validated correlation coefficient, indicating the goodness-of-fit. A value > 0.6 is required [4].
- r²pred (r²pred): The predictive correlation coefficient for an external test set. A value > 0.5 indicates good external predictive ability [4].
- Optimal Number of Components (ONC): The number of latent variables from the PLS analysis that yields the highest q² value [4].
- Field Contributions: The relative contribution of each field (e.g., steric, electrostatic) to the final model, which helps validate the chemical intuition [19].
Benchmarking with the Steroid Dataset
The original CoMSIA publication [19] used a steroid benchmark dataset to validate the method. Reproducing this study with tools like Py-CoMSIA using standard probe and grid parameters (e.g., grid spacing of 1 Å, padding of 4 Å, attenuation factor of 0.3) should yield comparable results to the original Sybyl implementation [19]. Successful benchmarking is indicated by:
- A cross-validated q² value comparable to the published result (e.g., ~0.665 for SEH fields) [19].
- Identification of similar key features in the contour maps.
- Comparable prediction of an external test set.
Table 4: Example Benchmark Results for Steroid Dataset (Py-CoMSIA vs. Sybyl)
| Metric | Published (SEH) | Py-CoMSIA (SEH) |
|---|---|---|
| q² | 0.665 | 0.609 |
| r² | 0.937 | 0.917 |
| SPRESS | 0.759 | 0.718 |
| No. Components | 4 | 3 |
| Steric Contribution | 0.073 | 0.149 |
| Electrostatic Contribution | 0.513 | 0.534 |
| Hydrophobic Contribution | 0.415 | 0.316 |
Source: Adapted from [19]
Troubleshooting and Advanced Optimization
Common Issues and Solutions
- Poor Statistical Results (low q²): This can stem from inadequate molecular alignment, incorrect bioactive conformation, or inappropriate grid placement. Employ All-Orientation Search (AOS) and All-Placement Search (APS) strategies to systematically test different rotations and translations of the molecular aggregate within the grid to find the orientation yielding the highest q² [78].
- Model Overfitting: The large number of grid points (variables) risks chance correlations. Apply variable selection algorithms (e.g., Genetic Algorithm, Enhanced Replacement Method) to filter out noisy variables and build simpler, more predictive models [78].
- Intermittent Results: Slight changes in grid placement can lead to different results, especially in CoMFA. Consider using the more stable CoMSIA method or the AOS/APS strategies for CoMFA [78].
Advanced Techniques
- Docking-Based Alignment: For targets with known 3D structure, using bioactive conformations extracted from molecular docking (e.g., using AutoDock or Glide) can provide a more physiologically relevant alignment for grid generation [80] [79].
- Consensus Modeling: Building multiple models with slightly different parameters (e.g., grid spacing, charge models) and using the consensus can improve predictive reliability.
- Integration with MD Simulations: The stability of binding modes inferred from 3D-QSAR contour maps can be confirmed using Molecular Dynamics (MD) simulations and binding free energy calculations (MM/PBSA) [79].
The following workflow outlines the comprehensive process from initial setup to final model validation and application, incorporating troubleshooting loops.
Handling Conformational Flexibility and Bioactive Conformer Selection
In the realm of 3D Quantitative Structure-Activity Relationship (3D-QSAR) studies, particularly those employing Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), the treatment of conformational flexibility and the selection of bioactive conformers represent a critical foundational step. The predictive power and robustness of the resulting models are highly contingent upon the accurate representation of the ligand geometry that interacts with the biological target [82] [83]. A fundamental challenge is that a small molecule in solution often exists as an ensemble of conformations, but only one, or a limited few, adopt the specific geometry—the bioactive conformation—upon binding to its receptor [83]. Incorrectly identifying this conformation introduces a significant source of error that subsequent analytical steps cannot rectify.
Traditional 3D-QSAR protocols often rely on a single, static alignment of molecules, sometimes derived from a crystal structure of a ligand-receptor complex or based on a postulated pharmacophore hypothesis [82]. However, for conformationally flexible molecules, identifying this alignment objectively is technically difficult and has been a bottleneck in the application of 3D-QSAR methods, discouraging their use in "real-world" drug discovery problems [82]. This application note details established and advanced protocols for handling conformational flexibility, ensuring that the generated CoMFA and CoMSIA models are both reliable and predictive.
Theoretical Background and Significance
The core principle underpinning the need for careful conformer selection is the complementarity between a ligand and its protein binding site. The 3D-QSAR approaches, like CoMFA and CoMSIA, function by correlating molecular fields—steric, electrostatic, hydrophobic, and hydrogen-bonding—around a set of aligned molecules with their biological activities [16] [4] [25]. If the molecules are not aligned in a geometry that reflects their binding mode, the resulting field calculations will be misaligned, leading to models with poor statistical quality and low predictive value [82] [83].
The sensitivity of 3D-QSAR to molecular alignment is well-documented. A model's explanatory and predictive power is directly linked to the "biological reality" of the chosen conformations and their relative orientations [82]. The conventional method of using a single, energy-minimized conformation as a "sophisticated guess" for the bioactive conformation is often an erroneous prerequisite [83]. This limitation has driven the development of more sophisticated, multi-conformer approaches, such as 4D-QSAR, which explicitly accounts for conformational flexibility, orientation, and protonation states by using an ensemble of molecular structures as the input for QSAR analysis [83].
Protocols for Conformer Selection and Alignment
Ligand-Based Alignment Using a Common Substructure
This protocol is most applicable to a congeneric series of compounds that share a common structural scaffold, such as a rigid steroid nucleus or a defined pharmacophore.
- Template Selection: Identify the most structurally common fragment across all molecules in the dataset. Often, the molecule with the highest biological activity is used as the primary template for alignment [16].
- Conformation Generation: For flexible molecules, generate a set of low-energy conformations. This can be done using a conformational import function available in molecular modeling suites (e.g., MOE), typically employing a molecular mechanics force field like MMFF94x and a solvation model to account for physiological conditions [82].
- Molecular Superimposition: Align all molecules onto the template by fitting their common substructure atoms. The field-fit method can be used, which minimizes the root-mean-square deviation (RMSD) by optimizing six rigid-body degrees of freedom, aiming to align the molecules based on their steric and electrostatic field similarities [16].
- Optimization (Optional): To account for the grid orientation dependency of CoMFA, an All Orientation Search (AOS) can be performed. This script systematically rotates the aligned molecular aggregate within the grid box and selects the orientation that yields the highest cross-validated correlation coefficient ((q^2)) [16].
Pharmacophore-Based Automated Alignment
For datasets with more structural diversity or when the bioactive conformation is unknown, a pharmacophore-based alignment is a powerful objective method. The software AutoGPA exemplifies this automated approach [82].
- Conformer Generation: Generate a comprehensive set of low-energy conformations for each molecule in the dataset [82].
- Pharmacophore Feature Assignment: Assign key pharmacophore features—such as hydrogen bond acceptors, hydrogen bond donors, hydrophobic areas, and charged centers—to each generated conformation [82].
- Pharmacophore Elucidation: Exhaustively search for common three-dimensional arrangements of these pharmacophore features (pharmacophore queries) across the most active molecules. This function identifies multiple plausible alignment hypotheses [82].
- Model Building and Scoring: For each pharmacophore query and its resulting molecular alignment, a 3D-QSAR model (e.g., CoMFA) is built. The alignments are then scored and ranked based on the predictive ability of their corresponding models, typically using the cross-validated (q^2) value. The model with the best (q^2) is selected [82].
Diagram: Automated Pharmacophore Alignment Workflow (AutoGPA)
Structure-Based Alignment from a Protein-Ligand Complex
When a high-resolution crystal structure of a ligand bound to the target protein is available, it provides the most direct information for alignment.
- Template Extraction: Obtain the bioactive conformation of a ligand (e.g., a known inhibitor) directly from the Protein Data Bank (PDB) file of the protein-ligand complex [82].
- Ligand Preparation: Isolate the ligand from the complex. If necessary, add hydrogen atoms and calculate partial atomic charges using an appropriate force field (e.g., Tripos force field, MMFF94x) [16] [82].
- Database Conformer Alignment: For each molecule in the dataset, generate low-energy conformers and select the one that can be most successfully superimposed onto the template ligand from the crystal structure, focusing on key functional groups or the common scaffold. Alternatively, dock molecules into the binding site and use the top-scoring pose for alignment.
Essential Reagents and Computational Tools
Table: The Scientist's Toolkit for Handling Conformational Flexibility
| Tool Category | Specific Software/Functions | Key Utility in Protocol |
|---|---|---|
| Molecular Modeling Suites | SYBYL (Tripos, Inc.) [16], MOE (Molecular Operating Environment) [82] | Provides integrated environment for sketching molecules, energy minimization, conformational search, molecular alignment, and running CoMFA/CoMSIA analyses. |
| Force Fields | Tripos Force Field [16], MMFF94x [82] | Used for geometry optimization and energy minimization of molecular structures to obtain stable, low-energy conformations. |
| Charge Calculation Methods | Gasteiger-Hückel Method [16] | Calculates partial atomic charges, which are critical for the accurate computation of electrostatic interaction fields in CoMFA. |
| Automated Alignment Software | AutoGPA [82] | Automates the process of pharmacophore identification, conformer selection, and molecular alignment, generating multiple 3D-QSAR models for objective selection. |
| Probe Atoms | sp3 Carbon (+1 charge) [16] [4] | Used to calculate steric (Lennard-Jones) and electrostatic (Coulombic) interaction energies at thousands of grid points around the molecules. |
Validation and Best Practices
Ensuring the selected conformations and alignments yield a robust model requires rigorous validation.
Statistical Validation: The model must be validated internally and externally. Key statistical parameters include:
- Internal Validation: Cross-validated correlation coefficient ((q^2)) should typically be > 0.5 [4].
- External Validation: Predictive correlation coefficient ((r^2_{pred})) should be > 0.5 [4].
- Other Metrics: Additional parameters like (r^2), RMSE, MAE, and (r^2_m) should meet established thresholds to avoid model overestimation [4].
Contour Map Analysis: Interpreting the 3D contour maps generated by CoMFA and CoMSIA is a form of qualitative validation. The maps should provide a chemically intuitive and rational explanation for the observed structure-activity relationships. For instance, a green steric contour near a region where bulky substituents increase activity validates the alignment [82] [4].
Sensitivity Analysis: Explore different alignment rules and conformational assumptions. The stability of the model's predictive performance across minor variations in alignment is a good indicator of its robustness [83].
Table: Key Statistical Metrics for Validating 3D-QSAR Models
| Metric | Formula/Description | Acceptance Threshold | Purpose |
|---|---|---|---|
| q² (LOO) | (q^2 = 1 - \frac{\sum{(y{exp} - y{pred})^2}}{\sum{(y{exp} - \bar{y}{train})^2}}) [4] | > 0.5 [4] | Internal predictive ability (Leave-One-Out cross-validation) |
| r² | Conventional correlation coefficient | > 0.6 [4] | Goodness-of-fit of the model |
| r²˅pred | (r^2_{pred} = 1 - \frac{PRESS}{SDEP}) [4] | > 0.5 [4] | External predictive ability on a test set |
| RMSE | Root Mean Square Error | As low as possible | Average error of prediction |
| ONC | Optimal Number of Components | Identified by highest q² [4] | Prevents model overfitting |
The handling of conformational flexibility is not a mere preliminary step but a foundational determinant of success in any 3D-QSAR study involving CoMFA or CoMSIA. While ligand-based alignment using a common substructure remains a valid approach for congeneric series, the development of automated, pharmacophore-based methods like AutoGPA has significantly advanced the field by providing an objective and robust solution for aligning flexible molecules in the absence of explicit receptor structural data [82]. The emerging trend of incorporating full-atom molecular dynamics simulations, as seen in 4D-QSAR approaches, promises a further leap by explicitly modeling the dynamic nature of ligand-receptor interactions [83]. By adhering to the detailed protocols and validation standards outlined in this application note, researchers can construct 3D-QSAR models with enhanced predictive power, thereby making more reliable decisions in the rational design of novel bioactive compounds.
Integrating 3D-QSAR with Molecular Docking for Enhanced Reliability
In modern computational drug discovery, 3D-QSAR and molecular docking have emerged as powerful complementary techniques. While 3D-QSAR models, particularly CoMFA and CoMSIA, excel at correlating the three-dimensional molecular fields of ligands with their biological activity, molecular docking provides critical insights into protein-ligand interactions and binding orientations [3]. The integration of these methods creates a synergistic workflow that significantly enhances the reliability and predictive power of virtual screening and lead optimization processes [3] [38]. This protocol details a robust framework for combining these approaches, enabling researchers to leverage the strengths of both methodologies while mitigating their individual limitations. The integrated approach has demonstrated success across multiple therapeutic targets, including kinase inhibitors for cancer therapy [10] [38] and inhibitors for neurodegenerative diseases [84] [85].
The combined 3D-QSAR and molecular docking methodology follows a sequential workflow where outputs from each stage inform subsequent steps. Alignment quality is crucial for 3D-QSAR model reliability, as misaligned molecules can generate statistically insignificant models [11]. Following 3D-QSAR model development and validation, the contour maps provide a visual guide for rational compound design, which can then be evaluated through molecular docking to assess potential binding modes and interactions [3] [38]. Molecular dynamics simulations further validate the stability of proposed ligand-receptor complexes [84] [38]. This multi-stage approach creates a feedback loop where insights from docking can refine 3D-QSAR models and vice versa, leading to more reliable predictions of biological activity and binding affinity.
Application Notes
Key Advantages of Integration
Integrating 3D-QSAR with molecular docking addresses critical limitations of using either method in isolation. The combination provides a more comprehensive assessment of potential drug candidates by evaluating both ligand-based and structure-based perspectives [3]. This integrated approach is particularly valuable for:
Overcoming 3D-QSAR Alignment Dependence: Molecular docking provides hypothesized bioactive conformations that can inform alignment strategies for 3D-QSAR, potentially improving model quality [38].
Validation of Binding Hypotheses: 3D-QSAR contour maps suggest favorable chemical modifications, which can be computationally validated through docking studies to assess whether these modifications improve complementary interactions with the target [3] [38].
Identification of Critical Interactions: Docking reveals specific protein-ligand interactions (hydrogen bonds, hydrophobic contacts, π-π stacking), while 3D-QSAR indicates which molecular features most significantly impact potency [10] [38].
Handling Target Flexibility: Molecular dynamics simulations following docking can account for receptor flexibility and provide insights into binding stability that static 3D-QSAR models cannot capture [84] [38].
Case Study: Bcr-Abl Inhibitor Design
A recent application of this integrated approach demonstrated its effectiveness in designing purine-based Bcr-Abl inhibitors for chronic myeloid leukemia treatment [10]. Researchers developed 3D-QSAR models using a dataset of 58 purine derivatives, with both CoMFA and CoMSIA models showing strong predictive capability. The contour maps guided the design of new compounds, which were then evaluated through docking studies against both wild-type and T315I mutant Bcr-Abl. This approach yielded compounds 7a and 7c with IC~50~ values of 0.13 and 0.19 μM respectively, surpassing imatinib's potency (IC~50~ = 0.33 μM) [10]. The integration of 3D-QSAR with docking facilitated the optimization of key substituents at the 2, 6, and 9 positions of the purine scaffold to maximize interactions with the binding pocket while maintaining favorable steric and electrostatic properties.
Case Study: MAO-B Inhibitor Development
In neurodegenerative disease research, an integrated 3D-QSAR and docking approach was applied to develop novel 6-hydroxybenzothiazole-2-carboxamide derivatives as monoamine oxidase B (MAO-B) inhibitors [84] [85]. The CoMSIA model demonstrated excellent statistical parameters (q² = 0.569, r² = 0.915), and contour map analysis identified favorable regions for steric bulk and electrostatic modifications. Docking studies revealed critical interactions with key residues in the MAO-B active site, enabling the design of compound 31.j3 which showed stable binding in molecular dynamics simulations with RMSD fluctuations between 1.0-2.0 Å [84]. This case highlights how the combined approach can optimize selective enzyme inhibitors by simultaneously considering ligand-based field contributions and structure-based interaction patterns.
Experimental Protocols
3D-QSAR Model Development Protocol
Data Set Preparation and Molecular Modeling
Compound Selection: Compile a structurally related but diverse set of compounds (typically 20-50 molecules) with consistently measured biological activity data (e.g., IC~50~, Ki) under uniform experimental conditions [11]. Divide compounds into training (~70-80%) and test sets (~20-30%) using random or structural diversity-based selection [38].
Structure Optimization: Convert 2D structures to 3D coordinates using tools like ChemDraw [84] or RDKit [11]. Perform geometry optimization using molecular mechanics (e.g., UFF) or quantum mechanical methods to obtain low-energy conformations [11].
Molecular Alignment: Align molecules using one of these approaches:
Descriptor Calculation and Model Building
Field Calculation: Calculate CoMFA steric (Lennard-Jones) and electrostatic (Coulombic) fields using a 3D grid with 2.0 Å spacing and a sp³ carbon probe with +1 charge [4]. For CoMSIA, additionally compute hydrophobic, hydrogen bond donor, and acceptor fields using Gaussian-type functions [11].
Partial Least Squares (PLS) Analysis: Build the QSAR model using PLS regression to correlate field descriptors with biological activity. Determine the optimal number of components using leave-one-out (LOO) cross-validation to maximize q² while minimizing overfitting [4] [38].
Model Validation
Internal Validation: Calculate LOO cross-validated correlation coefficient (q²) using:
External Validation: Evaluate predictive power using test set compounds:
Additional Validation Metrics: Assess model robustness using:
Table 1: Statistical Parameters for Validated 3D-QSAR Models from Case Studies
| Case Study | Method | q² | r² | r²~pred~ | Components | Field Contributions |
|---|---|---|---|---|---|---|
| Bcr-Abl Inhibitors [10] | CoMFA/CoMSIA | >0.5 | >0.6 | >0.5 | Not specified | Steric, Electrostatic |
| MAO-B Inhibitors [84] | CoMSIA | 0.569 | 0.915 | Not specified | Not specified | SEA* |
| TTK Inhibitors [38] | CoMFA | 0.583 | Not specified | 0.751 | Not specified | Steric, Electrostatic |
| TTK Inhibitors [38] | CoMSIA | 0.690 | Not specified | 0.767 | Not specified | Steric, Electrostatic, HBA, HBD, Hydrophobic |
*SEA: Steric, Electrostatic, Hydrogen bond Acceptor
Integrated Docking Protocol
System Preparation
Protein Preparation: Obtain 3D protein structure from PDB. Add missing hydrogen atoms, assign bond orders, and correct protonation states of residues using tools like Maestro Protein Preparation Wizard [38]. Perform energy minimization to relieve steric clashes.
Ligand Preparation: Generate 3D structures of newly designed compounds from 3D-QSAR contour maps. Optimize geometries using molecular mechanics and assign appropriate charges (MMFF94, Gasteiger-Hückel) [38].
Binding Site Definition: Define the binding site using known ligand coordinates from crystallographic data or through computational site detection methods.
Molecular Docking and Analysis
Docking Execution: Perform docking simulations using programs like AutoDock, GOLD, or Glide. Apply appropriate search algorithms and scoring functions [10] [38].
Pose Analysis and Selection: Cluster docking poses based on RMSD. Select representative poses that:
- Show consistency with 3D-QSAR contour maps
- Form key interactions with binding site residues
- Occupy the appropriate subpockets identified in structure-activity relationships
Interaction Analysis: Identify critical hydrogen bonds, hydrophobic contacts, π-π stacking, and salt bridges that contribute to binding affinity and specificity.
Molecular Dynamics Validation Protocol
System Setup: Solvate the protein-ligand complex in an appropriate water model (TIP3P). Add counterions to neutralize system charge. Employ periodic boundary conditions [84] [38].
Simulation Parameters: Perform energy minimization followed by gradual heating to 300K. Conduct production MD simulations for 50-100 ns using packages like AMBER, GROMACS, or Desmond [84] [38].
Trajectory Analysis: Calculate:
Binding Free Energy Calculations: Employ MM-PBSA or MM-GBSA methods to compute binding free energies and identify key residue contributions [46] [38].
Table 2: Key Validation Metrics from MD Simulations in Case Studies
| Case Study | Simulation Time | Complex Stability (RMSD) | Key Interactions | Binding Free Energy |
|---|---|---|---|---|
| MAO-B Inhibitors [84] | Not specified | 1.0-2.0 Å fluctuations | Van der Waals, Electrostatic | Not specified |
| Anti-breast Cancer Agents [46] | 100 ns | Stable after equilibration | H-bonds, Hydrophobic | MM-PBSA calculated |
| TTK Inhibitors [38] | Not specified | Stable complexes | Specific interactions with catalytic residues | Favorable MM/PBSA results |
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Category | Specific Tools/Reagents | Function/Purpose |
|---|---|---|
| Software Packages | Sybyl-X [84] [11], Schrödinger Suite [38], AutoDock, GROMACS/AMBER [38] | Integrated molecular modeling, 3D-QSAR, docking, and MD simulations |
| 3D-QSAR Specific | CoMFA, CoMSIA [3] [11] | Calculate steric, electrostatic, hydrophobic, and H-bond fields; generate contour maps |
| Validation Tools | Bootstrapping scripts, Statistical metrics (q², r²~pred~, r²~m~) [4] [38] | Assess model robustness, predictive power, and reliability |
| Structure Preparation | RDKit [11], ChemDraw [84], Protein Preparation Wizard [38] | Generate 3D coordinates, optimize geometries, prepare protein structures |
| Analysis & Visualization | Maestro [38], PyMOL, VMD | Analyze docking poses, MD trajectories, and contour maps |
The integration of 3D-QSAR with molecular docking represents a powerful paradigm in structure-based drug design that significantly enhances prediction reliability. This protocol provides a comprehensive framework that leverages the complementary strengths of both approaches: 3D-QSAR's ability to quantify structure-activity relationships through molecular field analysis, and molecular docking's capacity to elucidate binding modes and specific protein-ligand interactions. The case studies across diverse therapeutic targets demonstrate that this integrated methodology can successfully guide the design of novel compounds with improved potency and selectivity. As computational power increases and algorithms evolve, this combined approach is poised to become even more central to efficient drug discovery pipelines, potentially reducing the time and cost associated with experimental screening while providing valuable mechanistic insights for lead optimization.
Ensuring Model Robustness and Predictive Power
In the field of computational drug design, 3D Quantitative Structure-Activity Relationship (3D-QSAR) methodologies, notably Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), are pivotal for elucidating the relationship between a molecule's three-dimensional structure and its biological activity [4] [7]. The predictive ability and reliability of these models are critically dependent on robust statistical validation [86]. Without proper validation, models risk overfitting, where a model learns the noise in the training data rather than the underlying relationship, leading to poor performance on new, unseen data [87] [88]. This article details the application of cross-validation and external test set validation within 3D-QSAR protocols, providing a structured guide for researchers and drug development professionals to develop predictive and trustworthy models.
Core Concepts and Definitions
The Need for Validation
The primary goal of a QSAR model is not just to explain the data it was built on, but to accurately forecast the activity of novel compounds [86]. A model's performance on its training data is often an optimistic estimate of its true predictive capability [87]. Validation techniques are therefore employed to estimate this generalization error [87] [89]. Key metrics used in these validation processes include the cross-validated correlation coefficient (q²)
for internal validation and the predictive correlation coefficient (r²pred)
for external validation [4].
Types of Validation
- Internal Validation: Assesses model stability and reliability using only the data used for model building. The most common method is cross-validation [4].
- External Validation: The gold standard for evaluating predictive ability, where the model's performance is tested on a completely independent set of compounds that were not used in any part of the model building process [86] [90].
Table 1: Key Validation Metrics and Their Thresholds for a Valid 3D-QSAR Model
| Metric | Description | Recommended Threshold |
|---|---|---|
q² |
Cross-validated correlation coefficient from internal validation | > 0.5 [4] [7] |
r² |
Non-cross-validated correlation coefficient of the training set | > 0.6 [4] |
r²pred |
Predictive correlation coefficient for the external test set | > 0.5 [4] |
n |
Optimal number of components | Should be reasonable to avoid overfitting [7] |
SEE |
Standard Error of Estimate | Should be as low as possible [7] |
F |
F-test value | Should be high, indicating model significance [7] |
Cross-Validation Methods and Protocols
Cross-validation is a resampling technique used to assess how a model will generalize to an independent dataset by partitioning the available data into training and validation sets multiple times [87] [88].
Common Cross-Validation Variants
- Leave-One-Out (LOO) CV: One compound is removed from the dataset and used as a validation set, while the remaining
N-1compounds are used as the training set. This is repeated until every compound has been left out once [87] [4]. LOO is useful for small datasets but can have high variance [87]. - k-Fold CV: The dataset is randomly partitioned into
kequal-sized folds. A model is trained onk-1folds and validated on the remaining fold. This process is repeatedktimes, with each fold used exactly once as the validation set [87] [88]. Stratified k-fold cross-validation ensures that each fold has a similar distribution of response values, which is crucial for datasets with imbalanced activities [87] [89]. - Double (Nested) Cross-Validation: An advanced technique consisting of two nested loops. The outer loop manages the data split into training and test sets, while the inner loop performs cross-validation on the training set for model selection and hyperparameter tuning [89] [90]. This method provides a nearly unbiased estimate of the true prediction error and is especially valuable under model uncertainty [90].
Experimental Protocol: Implementing k-Fold Cross-Validation in 3D-QSAR
Objective: To perform a robust internal validation of a 3D-QSAR model using 10-fold cross-validation.
Materials:
- A dataset of
Ncompounds with known biological activities (e.g., pIC₅₀ values). - Molecular modeling software (e.g., SYBYL, Open3DALIGN, or similar).
- A computer with a 3D-QSAR program and statistical analysis capabilities.
Procedure:
- Data Preparation: Prepare and align your set of
Nmolecules according to your standard 3D-QSAR protocol [4]. - Random Splitting: Randomly shuffle the dataset and divide it into
k=10mutually exclusive folds of approximately equal size. - Model Training and Validation:
- For
i = 1tok:- Set aside fold
ias the temporary validation set. - Use the remaining
k-1folds (9 folds) as the training set to build a CoMFA or CoMSIA model. - Use the generated model to predict the biological activity of the compounds in the validation fold
i. - Calculate the squared difference between the predicted and experimental activity for each compound in the validation fold.
- Set aside fold
- For
- Calculation of
q²:- After all
kcycles, combine all predictions from each validation fold. - Calculate the Predictive Residual Sum of Squares (PRESS):
PRESS = Σ(y_actual - y_predicted)² - Calculate the Total Sum of Squares (SS):
SS = Σ(y_actual - y_mean)² - Compute the cross-validated coefficient
q²:q² = 1 - (PRESS / SS)
- After all
- Interpretation: A
q²value greater than 0.5 is generally considered indicative of a robust model [4] [7].
Figure 1: A 10-Fold Cross-Validation Workflow for 3D-QSAR Model Validation.
External Validation with a Test Set
While cross-validation is an essential internal validation step, it is not a substitute for external validation [86]. External validation provides the most rigorous assessment of a model's predictive power.
Experimental Protocol: External Validation for a CoMFA/CoMSIA Model
Objective: To evaluate the true predictive performance of a finalized 3D-QSAR model on a set of compounds that were entirely excluded from the model building process.
Materials:
- The full dataset of
Ncompounds. - A finalized 3D-QSAR model (e.g., a CoMFA model with defined fields and coefficients).
Procedure:
- Initial Data Division: Before any model building, randomly divide the entire dataset into a training set (typically 80-90%) and a test set (10-20%) [88] [89]. The test set must be set aside and not used in any way during model development or optimization.
- Model Building: Build the final CoMFA/CoMSIA model using only the training set compounds. This includes all steps: alignment, field calculation, and PLS regression.
- Prediction: Use the finalized model to predict the biological activities of the compounds in the external test set.
- Calculation of
r²pred:- Calculate the PRedictive Error Sum of Squares (PRESS) for the test set:
PRESS_test = Σ(y_actual,test - y_predicted,test)² - Calculate the Total Sum of Squares (SS) for the test set based on the mean activity of the training set:
SS_test = Σ(y_actual,test - y_mean,training)² - Compute the predictive
r²pred:r²pred = 1 - (PRESS_test / SS_test)
- Calculate the PRedictive Error Sum of Squares (PRESS) for the test set:
- Interpretation: An
r²predgreater than 0.5 is a key indicator that the model has satisfactory predictive ability [4].
Table 2: Comparison of Common Validation Strategies in QSAR
| Strategy | Procedure | Key Metric(s) | Advantages | Disadvantages |
|---|---|---|---|---|
| Hold-Out | Single split into training and test sets. | r²pred |
Simple and fast [88]. | Estimate can be highly dependent on a single, fortuitous split; inefficient data use [87] [90]. |
| k-Fold CV | Data split into k folds; each fold serves as a validation set once. | q² |
More reliable than hold-out; uses data more efficiently [88]. | Can be computationally expensive; estimates can have high variance if k is too large [87]. |
| LOO CV | k = N; each compound is left out once. | q² |
Low bias; ideal for very small datasets [87]. | High variance; can lead to overoptimistic estimates in case of data clustering [91] [87]. |
| Double CV | Nested loops for model selection and error estimation. | q² (outer loop) |
Minimizes model selection bias; provides realistic error estimation [90]. | Computationally very intensive [89] [90]. |
A Comprehensive Validation Workflow: Integrating Cross-Validation and External Testing
For the most robust assessment, a combination of internal and external validation should be employed. The following workflow integrates double cross-validation with a final external test.
Figure 2: A Comprehensive Workflow Integrating Double Cross-Validation and an External Test Set.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for 3D-QSAR Validation
| Reagent / Tool | Function / Description | Application in Protocol |
|---|---|---|
| Molecular Dataset | A curated set of compounds with reliable and consistent biological activity data (e.g., IC₅₀). | The foundational input for all model building and validation [5] [7]. |
| Alignment Rule | A consistent method for superimposing molecules based on a common scaffold or pharmacophore. | Critical for generating meaningful 3D molecular fields in CoMFA/CoMSIA [4] [7]. |
| 3D-QSAR Software | Software capable of calculating steric, electrostatic, and other molecular fields (e.g., SYBYL, Open3DQSAR). | Used to compute interaction energies and build the PLS regression models [4] [7]. |
| Statistical Software/Package | A tool for performing statistical analysis and cross-validation (e.g., scikit-learn, R, built-in QSAR software modules). | Used to implement k-fold splits, calculate q², r²pred, and other validation metrics [88] [90]. |
| Validation Scripts | Custom or pre-written scripts (e.g., in Python) to automate double cross-validation and metric calculation. | Ensures reproducibility and reduces human error in complex validation procedures like double CV [89] [90]. |
Robust statistical validation is the cornerstone of developing reliable and predictive 3D-QSAR models. The integration of internal cross-validation, to guide model selection and ensure robustness, with a final external test set validation, to provide an unbiased estimate of predictive power, is paramount [86] [90]. By adhering to the detailed protocols and workflows outlined in this application note, researchers can confidently validate their CoMFA and CoMSIA models, thereby accelerating the rational design of novel bioactive compounds in drug development.
Interpreting Contour Maps for Rational Drug Design
In rational drug design, the biological activity of a ligand is determined by its three-dimensional interaction with a biological receptor. The receptor perceives the ligand not as a set of atoms, but as a shape carrying complex molecular forces [2]. Three-dimensional Quantitative Structure-Activity Relationship (3D-QSAR) methods, particularly Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), model these interactions by calculating the molecular fields around a set of aligned compounds. The results are visualized as contour maps that indicate regions where specific chemical features enhance or diminish biological activity [11] [2]. These maps provide a visual and quantitative guide for medicinal chemists to optimize molecular structures, making them indispensable in modern drug discovery campaigns, especially when the detailed structure of the target receptor is unknown [2].
Fundamental Principles of Contour Map Interpretation
The Conceptual Foundation of Molecular Fields
The core principle of 3D-QSAR is that the biological activity of a compound correlates with the steric and electrostatic fields it presents to the receptor [2]. These fields are sampled by placing a probe atom at thousands of points on a grid that surrounds the aligned molecules [2]. The steric field is typically probed with an sp³ carbon atom and calculated using a Lennard-Jones potential, representing van der Waals interactions. The electrostatic field is probed with a charged sp³ carbon atom and calculated using Coulomb's law [73] [2]. Statistical methods, primarily Partial Least Squares (PLS) regression, are then used to correlate the variations in these field values with the variations in biological activity across the compound set, resulting in a predictive model [11].
Decoding the Contour Map Colors and Regions
Contour maps translate the statistical model into an intuitive, visual format by highlighting critical regions in 3D space around a reference molecule. The interpretation of these maps is standardized by color codes and their associated meanings, summarized in the table below.
Table 1: Standard Contour Map Color Interpretation in CoMFA and CoMSIA
| Field Type | Color | Structural Implication |
|---|---|---|
| Steric | Green | Bulky groups in this region increase activity. |
| Yellow | Bulky groups in this region decrease activity. | |
| Electrostatic | Blue | Positively charged groups increase activity. |
| Red | Negatively charged groups increase activity. | |
| Hydrophobic | Yellow | Hydrophobic groups increase activity [92]. |
| White | Hydrophobic groups decrease activity [92]. | |
| Hydrogen Bond Donor | Cyan | Hydrogen bond donor groups increase activity [92]. |
| Purple | Hydrogen bond donor groups decrease activity [92]. |
For example, a green contour indicates a region where increasing molecular bulk (e.g., changing a hydrogen to a methyl group) is favorable for activity, likely by filling a hydrophobic pocket in the protein. Conversely, a yellow steric contour warns of a potential clash with the receptor. A blue electrostatic contour suggests the receptor environment is favorable for a positive charge, guiding the chemist to introduce or maintain a basic group in that area [11].
Experimental Protocol for Generating and Applying Contour Maps
The following workflow outlines the standard procedure for conducting a 3D-QSAR study, from data preparation to the application of contour maps for drug design.
Diagram 1: 3D-QSAR Workflow for Drug Design
Data Collection and Preparation
The first step involves assembling a dataset of compounds with experimentally determined biological activities (e.g., IC₅₀, Ki) measured under uniform conditions [11]. Typically, 20-50 structurally related but diverse compounds are required. This dataset is divided into a training set for model building and a test set for external validation [93]. The predictive quality of the final model is highly dependent on the quality and consistency of this initial data.
Molecular Modeling and Alignment
- 3D Structure Generation: Two-dimensional molecular structures are converted into three-dimensional coordinates using tools like RDKit or Sybyl and then geometry-optimized to low-energy conformations using molecular mechanics (e.g., Universal Force Field) or quantum mechanical methods [11].
- Molecular Alignment: This is a critical and sensitive step. All molecules must be superimposed in a way that reflects their common binding mode at the target site [11] [94]. Common alignment methods include:
- Pharmacophore-Based Alignment: Using a known active compound or a common substructure as a template.
- Database Alignment: Superimposing molecules based on the largest common substructure shared across the dataset [11].
- Protein-Based Alignment: If the protein structure is available, ligands can be aligned based on their docked poses within the binding site [94].
Field Calculation and Model Building
- Descriptor Calculation: For CoMFA, steric (Lennard-Jones) and electrostatic (Coulomb) fields are calculated at each grid point using a probe atom [73]. For CoMSIA, which often provides smoother and more interpretable maps, Gaussian-type functions are used to calculate similarity indices for steric, electrostatic, hydrophobic, and hydrogen bond donor/acceptor fields [11] [94].
- Model Construction and Validation: Partial Least Squares (PLS) regression is used to correlate the field descriptors with biological activity [11]. The model is validated using:
- Cross-Validation: Leave-one-out (LOO) cross-validation yields the q² value, where q² > 0.5 is generally considered predictive [10] [93].
- External Validation: Predicting the activity of the test set compounds provides the r²ₚᵣₑ𝒹 value, which is a crucial indicator of the model's robustness and predictive power for new compounds [93].
Contour Map Interpretation and Analog Design
Once a statistically valid model is obtained, the contour maps are generated and superimposed on a reference molecule. The medicinal chemist analyzes these maps to identify specific structural modifications. For instance, a green steric contour near a substituent suggests that enlarging that group could improve potency, while a red electrostatic contour near a phenyl ring might suggest introducing an electron-withdrawing group to enhance activity [11].
Research Reagent Solutions
The following table details key software and computational tools required for conducting 3D-QSAR studies.
Table 2: Essential Research Reagents and Software for 3D-QSAR
| Tool/Reagent | Function in 3D-QSAR | Specific Application Example |
|---|---|---|
| SYBYL (Tripos) | Integrated molecular modeling suite. | Industry-standard software for performing CoMFA and CoMSIA studies [73] [94] [93]. |
| RDKit | Open-source cheminformatics toolkit. | Generating 3D conformers and performing molecular alignment [11]. |
| PLS Algorithm | Statistical correlation method. | Core algorithm in SYBYL for building the relationship between molecular fields and biological activity [11]. |
| Probe Atoms | Calculate molecular interaction fields. | sp³ C+1 charge for electrostatic fields; sp³ C for steric fields [73] [2]. |
| Grid Box | 3D lattice for spatial sampling. | Defines the region around aligned molecules where field values are calculated [2]. |
Application in Rational Drug Design: Case Studies
Overcoming Multidrug Resistance in Cancer
A prominent application involved designing inhibitors for the Multidrug Resistance Protein 1 (MRP1), an efflux pump that confers resistance to chemotherapeutic agents. 3D-QSAR studies on a series of tariquidar analogues established highly predictive CoMFA (r² = 0.968) and CoMSIA (r² = 0.982) models [92]. The resulting contour maps demonstrated that steric, electrostatic, hydrophobic, and hydrogen bond donor fields were critical for MRP1 inhibition. These maps provided a structural rationale for designing novel, potent, and selective MDR modulators, guiding optimizations to specific regions of the tariquidar scaffold to block the efflux of anti-cancer drugs effectively [92].
Designing Novel Bcr-Abl Inhibitors for Leukemia
In the fight against Chronic Myeloid Leukemia (CML), resistance to imatinib due to Bcr-Abl mutations is a major challenge. 3D-QSAR was successfully employed to design new purine-based Bcr-Abl inhibitors [10]. The study used a dataset of 58 purine derivatives to build CoMFA and CoMSIA models. The contour maps guided the design of new substituents at the 2, 6, and 9 positions of the purine core. This led to the synthesis of compound 7c, which exhibited superior potency (IC₅₀ = 0.19 µM) compared to imatinib (IC₅₀ = 0.33 µM) and was also effective against resistant cell lines, showcasing the power of 3D-QSAR in addressing drug resistance [10].
Development of Non-Covalent Proteasome Inhibitors
In a study targeting the 20S proteasome, researchers developed robust CoMFA and CoMSIA models for a series of phenol ether derivatives [93]. The best models showed high predictive power (CoMFA r²ₚᵣₑ𝒹 = 0.755; CoMSIA r²ₚᵣₑ𝒹 = 0.921). Analysis of the contour maps provided critical clues for structural optimization, leading to the design of 24 novel non-covalent inhibitors. Molecular docking suggested that the high activity of the newly designed compounds was due to optimal hydrogen bonding and hydrophobic interactions within the proteasome's binding pocket, insights initially derived from the contour maps [93].
Comparing CoMFA and CoMSIA Model Performance
Within the domain of three-dimensional quantitative structure-activity relationship (3D-QSAR) modeling, Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) represent two pivotal methodological approaches. Both techniques are foundational to rational drug design, enabling researchers to correlate the three-dimensional structural properties of compounds with their biological activities. This application note provides a detailed comparison of CoMFA and CoMSIA model performance, framed within the context of establishing robust 3D-QSAR protocols. The content is structured to guide researchers, scientists, and drug development professionals in selecting and implementing the appropriate methodology for their specific research challenges, with a focus on practical application and interpretability of results.
Theoretical Foundations and Comparative Descriptors
The core distinction between CoMFA and CoMSIA lies in their fundamental approaches to describing molecular fields. CoMFA, the earlier developed method, calculates steric and electrostatic fields based on Lennard-Jones and Coulombic potentials, respectively [16] [12]. It employs a probe atom placed at grid points to measure interaction energies, which are often truncated at predefined energy cutoffs (e.g., 30 kcal/mol) to avoid unrealistic values [16] [12]. This approach can result in abrupt, discontinuous field distributions near molecular surfaces.
In contrast, CoMSIA introduces a Gaussian-type function to calculate similarity indices, which avoids sharp energy cutoffs and generates continuous molecular similarity maps [19] [69]. This methodological advancement makes CoMSIA models less sensitive to molecular alignment and grid spacing parameters compared to CoMFA [19] [69]. Furthermore, CoMSIA extends the analytical scope by incorporating up to five distinct molecular fields: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor [19] [25] [71]. This provides a more holistic view of the molecular determinants underlying biological activity, particularly in cases where hydrophobic forces or hydrogen bonding dominate receptor-ligand recognition.
Table 1: Fundamental Comparison of CoMFA and CoMSIA Descriptors
| Feature | CoMFA | CoMSIA |
|---|---|---|
| Field Types | Steric, Electrostatic | Steric, Electrostatic, Hydrophobic, H-Bond Donor, H-Bond Acceptor |
| Calculation Method | Lennard-Jones & Coulomb potentials | Gaussian-type distance dependence |
| Field Distribution | Discontinuous near molecular surface | Continuous |
| Sensitivity to Alignment | Relatively higher | Less sensitive |
| Probe Atom | sp³ carbon, +1 charge [16] | sp³ carbon, +1 charge, +1 hydrophobicity, +1 H-bond properties [16] |
| Standard Attenuation Factor | Not applicable | 0.3 [19] [16] |
Performance Benchmarking and Statistical Validation
Predictive Performance Across Diverse Datasets
Independent benchmarking studies across various chemical datasets provide practical insights into the comparative performance of CoMFA and CoMSIA. A comprehensive evaluation on eight Sutherland datasets revealed that while both methods show variable performance depending on the specific biological target, CoMFA often demonstrates a slight edge in predictive capability [95].
Table 2: Comparative Model Performance Across Benchmarking Studies
| Dataset/Target | CoMFA (COD) | CoMSIA Basic (COD) | CoMSIA Extra (COD) | Reference |
|---|---|---|---|---|
| BACE-1 Inhibitors | 0.33 | 0.13 | Not reported | [95] |
| ACE | 0.49 | 0.52 | 0.49 | [95] |
| ACHE | 0.47 | 0.44 | 0.44 | [95] |
| BZR | 0.0 | 0.08 | 0.12 | [95] |
| COX2 | 0.29 | 0.03 | 0.37 | [95] |
| Steroids (q²) | 0.665 (Sybyl) | 0.609 (Py-CoMSIA SEH) | 0.630 (Py-CoMSIA SEHAD) | [19] |
The performance variation highlights the importance of field selection in CoMSIA. For instance, in the steroid benchmark test case, CoMSIA with steric, electrostatic, and hydrophobic (SEH) fields yielded a cross-validated q² of 0.609, comparable to Sybyl's CoMSIA result of 0.665 [19]. However, incorporating all five fields (SEHAD) showed somewhat reduced predictive capacity (r²pred = 0.186 for SEHAD vs. 0.319 for SEH) [19], suggesting that additional fields do not universally guarantee improved performance and must be selected judiciously based on the specific receptor-ligand interaction context.
Model Validation Protocols
Robust validation is paramount for reliable QSAR models. External validation remains the gold standard for assessing predictive capability [96]. The following statistical parameters should be routinely reported:
- q²: Cross-validated correlation coefficient (should be > 0.5 for a predictive model)
- r²: Non-cross-validated correlation coefficient
- r²pred: Predictive r² for external test set (should be > 0.6) [96]
- Standard Error of Estimate
- Optimal Number of Components
- Field Contributions
More sophisticated validation approaches include the Golbraikh and Tropsha criteria [96] and the concordance correlation coefficient (CCC), which should exceed 0.8 for a valid model [96]. Researchers should avoid relying solely on r² for model validation, as it alone cannot sufficiently indicate model validity [96].
Experimental Protocols and Workflow
Standardized CoMFA/CoMSIA Protocol
The following workflow outlines a standardized protocol for conducting CoMFA and CoMSIA studies, compiled from multiple experimental procedures [16] [71] [12]:
Critical Protocol Parameters
Successful implementation requires careful attention to these critical parameters:
- Molecular Alignment: The most crucial step. Use pharmacophore-based alignment (e.g., GALAHAD) for structurally diverse compounds or common substructure alignment for congeneric series [12].
- Grid Spacing: Typically 1.0-2.0 Å. Smaller spacing increases computational load but may capture finer details [16] [71].
- Attenuation Factor: For CoMSIA, the default value of 0.3 is generally effective [19] [16].
- Column Filtering: Reduces noise by omitting columns with small energy variance (typically 2.0 kcal/mol, but sometimes lowered to 0.3 kcal/mol for enhanced signal) [16].
- Region Focusing: In CoMFA, this technique can be applied to improve weight for lattice points most relevant to the model [16].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Essential Resources for 3D-QSAR Studies
| Resource Category | Specific Tools | Application in CoMFA/CoMSIA |
|---|---|---|
| Commercial Software | SYBYL (Tripos) [16] [12], Schrödinger, MOE | Traditional platforms offering comprehensive CoMFA/CoMSIA implementations with graphical interfaces |
| Open-Source Alternatives | Py-CoMSIA [19] [69], Open3DQSAR [95] | Python-based implementations increasing accessibility and flexibility for method customization |
| Molecular Descriptors | CoMFA Steric/Elec. fields, CoMSIA similarity indices (5 fields) | Field calculation specific to each method as described in Table 1 |
| Statistical Analysis | Partial Least Squares (PLS) regression [12] | Correlating field values with biological activity to generate predictive models |
| Validation Tools | Golbraikh-Tropsha criteria [96], CCC, rm² metrics | Assessing model robustness and predictive capability for external compounds |
The choice between CoMFA and CoMSIA should be guided by specific research objectives and the nature of the molecular system under investigation. CoMFA often provides slightly better predictive performance for systems where steric and electrostatic interactions dominate, while CoMSIA offers more comprehensive interaction profiling, particularly when hydrophobic or hydrogen bonding interactions are crucial.
For novel research, begin with CoMSIA using all five fields to identify which interaction types contribute most significantly to biological activity. For optimization campaigns focused on well-understood scaffolds, CoMFA or targeted CoMSIA with specific field combinations may yield more interpretable results. The emergence of open-source implementations like Py-CoMSIA [19] [69] now makes these powerful techniques more accessible, while commercial packages continue to offer refined workflows and support.
Regardless of the method selected, rigorous validation using both internal and external datasets remains paramount. Adherence to the standardized protocols outlined in this application note will ensure the development of robust, predictive 3D-QSAR models that can effectively guide drug discovery and optimization efforts.
Quantitative Structure-Activity Relationship (QSAR) modeling constitutes a cornerstone of modern computational drug discovery, enabling the prediction of biological activity from molecular structure. The field has evolved significantly from classical statistical methods to incorporate advanced artificial intelligence (AI) and deep learning techniques [97]. Among these advancements, Convolutional Neural Networks (CNNs) have emerged as a powerful tool for handling complex molecular data, particularly when integrated with established multi-dimensional QSAR frameworks like Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) [98] [99]. These 3D-QSAR methods traditionally correlate biological activity with non-covalent interaction fields—steric, electrostatic, hydrophobic, and hydrogen-bonding—surrounding a set of aligned molecules [4] [25]. The integration of CNNs enhances this paradigm by automatically extracting critical spatial features from molecular grids, leading to models with superior predictive power and robustness, especially in data-scarce scenarios common to lead optimization [98]. This Application Note details the protocols and recent advances in CNN-based 3D-QSAR, providing a structured guide for researchers aiming to implement these cutting-edge methodologies.
Key Advances in CNN-Integrated 3D-QSAR
The convergence of CNN architectures with traditional 3D-QSAR principles has led to the development of novel methodologies and tangible improvements in predictive performance. Table 1 summarizes the core characteristics and validation metrics of prominent approaches.
Table 1: Comparison of Traditional 3D-QSAR and Advanced CNN-Based Approaches
| Methodology | Core Description | Key Advantages | Reported Performance Metrics | Primary Application Context |
|---|---|---|---|---|
| CoMFA [4] [94] | Correlates steric and electrostatic molecular fields with biological activity using PLS. | High interpretability via 3D contour maps. | ( q^2 > 0.5 ), ( R^2 > 0.9 ) common in robust models [94]. | Lead optimization for enzyme inhibitors (e.g., HIV-1 PR) [94]. |
| CoMSIA [4] [25] | Extends CoMFA by incorporating hydrophobic and H-bond donor/acceptor fields. | Provides a more holistic view of interactions; avoids singularities. | ( q^2 ) up to 0.719 reported [100]. | Understanding multifaceted ligand-target interactions. |
| L3D-PLS [98] | CNN module extracts features from molecular grids, followed by PLS regression. | Superior predictive accuracy over CoMFA; automated feature extraction. | Outperformed CoMFA in 30 public molecular datasets [98]. | Ligand-based virtual screening without target protein structure. |
| CNN-QSAR for Cardiotoxicity [99] | CNN model trained on molecular descriptors to predict hERG channel blockade. | High predictive accuracy for a critical toxicity endpoint. | Training: ( Q^2 = 0.99 ), Test: ( R^2 = 0.70 ) [99]. | Early-stage prediction of cardiotoxicity risk. |
A landmark advancement is the L3D-PLS method, which replaces the manual feature engineering of traditional 3D-QSAR with an automated CNN feature extractor. The process involves creating 3D grids around pre-aligned ligand molecules, from which the CNN learns spatially invariant interaction patterns [98]. This approach has demonstrated statistically significant outperformance over traditional CoMFA across diverse, publicly available molecular datasets, highlighting its robustness and generalizability [98].
In parallel, CNN models have shown exceptional proficiency in predicting specific, complex biological endpoints such as cardiotoxicity mediated by the hERG potassium channel. These models achieve remarkably high correlation coefficients (( Q^2 = 0.99 )) on training data and maintain good predictive power (( R^2 = 0.70 )) on test data, providing a reliable tool for de-risking drug candidates early in development [99].
Experimental Protocols
Protocol 1: Building a Traditional 3D-QSAR Model (CoMFA/CoMSIA)
This protocol outlines the foundational steps for creating robust CoMFA and CoMSIA models, which serve as a benchmark for newer methods.
Workflow Overview:
Step-by-Step Procedure:
Dataset Curation and Preparation: Assemble a congeneric series of compounds (typically >20) with consistent biological activity data (e.g., IC50, Ki). Convert concentrations to pIC50 (-logIC50) to ensure a linear distribution [4] [101]. Divide the dataset randomly into a training set (~75-85%) for model building and a test set (~15-25%) for external validation [4] [101].
Molecular Alignment and Conformational Analysis: This is the most critical step for model success. Select a template molecule, typically the most active or rigid one. Align all molecules based on a common scaffold or pharmacophore using the database alignment method [94]. The quality of alignment directly dictates the robustness and predictivity of the final model [4].
Field Calculation:
- Place the aligned molecules into a 3D grid with a spacing of 2.0 Å [4].
- For CoMFA, calculate steric (Lennard-Jones potential) and electrostatic (Coulombic potential) fields using an sp3 carbon atom with a +1 charge as the probe [4].
- For CoMSIA, calculate up to five fields: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor. A common probe has +1 charge, +1 hydrophobicity, +1 donor, and +1 acceptor properties [4] [25].
Partial Least Squares (PLS) Regression Analysis: Use the PLS algorithm to correlate the calculated field values (independent variables) with the biological activity (dependent variable). Perform Leave-One-Out (LOO) cross-validation to determine the optimal number of components (ONC) and obtain the cross-validated correlation coefficient ( q^2 ) [4] [94]. A ( q^2 > 0.5 ) is generally considered a indicator of a robust model [4].
Model Validation: Validate the model using both internal and external validation techniques [4] [101].
- Internal Validation: Use the training set and LOO cross-validation.
- External Validation: Predict the activity of the test set compounds, which were not used in model building. Calculate the predictive correlation coefficient ( r^2_{pred} ) which should be >0.5 [4].
- Additional Metrics: Use other metrics like ( r^2_m ), RMSE (Root Mean Square Error), and MAE (Mean Absolute Error) to comprehensively assess model quality [4].
Interpretation via Contour Maps: Generate StDev*Coeff contour maps to visualize regions in 3D space where specific molecular properties favor or disfavor biological activity. These maps are crucial for guiding the rational design of new compounds [4] [94].
Protocol 2: Developing a CNN-Based 3D-QSAR Model (L3D-PLS)
This protocol describes the modern approach of integrating CNNs for enhanced feature learning in 3D-QSAR.
Workflow Overview:
Step-by-Step Procedure:
Input Data Generation: Start with a set of pre-aligned molecular structures, as in traditional 3D-QSAR. Represent each molecule as a 3D grid (e.g., 20x20x20 Å). Each voxel in the grid stores interaction energy values (e.g., steric, electrostatic) computed using a probe atom, creating multi-channel 3D images of the molecules [98].
CNN Architecture and Feature Extraction: Design a CNN module to process the 3D grids. A typical architecture includes:
- Convolutional Layers: Apply 3D convolutional filters to scan the input grids. These layers learn to detect local spatial features, such as steric bulks or electrostatic potential wells, that are critical for binding.
- Pooling Layers: Use max-pooling to reduce spatial dimensionality, ensuring translation invariance and controlling overfitting.
- Flattening: The final feature maps are flattened into a 1D feature vector. This vector represents the high-level, abstract features automatically extracted by the CNN from the molecular fields [98] [99].
Activity Prediction:
- The extracted feature vector is fed into a prediction layer. In the L3D-PLS framework, a Partial Least Squares (PLS) model is used to regress these features against the biological activity data [98].
- Alternatively, the feature vector can be connected to a fully connected (dense) neural network layer for a fully deep learning-based regression [99].
Model Training and Validation: Train the entire model (CNN + predictor) using the training set. Monitor performance on a separate validation set to avoid overfitting. Finally, evaluate the model's predictive power on the held-out test set using standard QSAR validation metrics (( R^2 ), ( q^2 ), RMSE, etc.), as detailed in Protocol 1 [98] [99].
The Scientist's Toolkit: Essential Research Reagents & Software
Successful implementation of 3D-QSAR studies relies on a suite of specialized software tools and computational resources. Table 2 lists key solutions for different stages of the workflow.
Table 2: Key Research Reagent Solutions for 3D-QSAR Modeling
| Tool/Resource Name | Type/Category | Primary Function in QSAR | Relevance to Protocol |
|---|---|---|---|
| SYBYL/Open3DALIGN [94] | Commercial & Open-Source Software | Molecular structure alignment and 3D-QSAR (CoMFA/CoMSIA) model generation. | Protocol 1 (Steps 2, 3, 4, 6) |
| PowerMV [99] | Descriptor Calculation Tool | Computes molecular descriptors and pharmacophore fingerprints for QSAR. | Protocol 2 (Step 1 - Descriptor calc.) |
| Python (Keras, PyTorch) [97] | Deep Learning Framework | Building, training, and validating custom CNN architectures for QSAR. | Protocol 2 (Steps 2, 3, 4) |
| QSARINS [97] | Standalone Software | Development and validation of QSAR models with extensive validation tools. | Protocol 1 (Steps 4, 5) |
| CORINA [99] | 3D Structure Generator | Converts 1D/2D molecular structures (e.g., SMILES) into 3D coordinate formats. | Protocol 1 & 2 (Step 1) |
| scikit-learn [97] | Python ML Library | Provides PLS regression, SVM, and other ML algorithms for model building. | Protocol 1 & 2 (Step 4) |
The integration of CNN-based approaches with multi-dimensional QSAR represents a significant leap forward in computational drug discovery. While traditional CoMFA and CoMSIA methods remain invaluable for their interpretability and well-established protocols, CNN-enhanced models like L3D-PLS offer demonstrably superior predictive accuracy by automating the extraction of critical features from 3D molecular space [98]. The protocols detailed herein provide a clear roadmap for researchers to implement both classical and state-of-the-art methods. As the field evolves, the synergy between explainable 3D-QSAR contours and the power of deep learning is poised to become the new standard, accelerating the efficient optimization of lead compounds and the identification of safer, more effective therapeutics [97].
Best Practices for Reporting and Applying 3D-QSAR Results
Three-dimensional Quantitative Structure-Activity Relationship (3D-QSAR) modeling represents a pivotal computational approach in modern drug discovery, enabling researchers to correlate the biological activity of compounds with their three-dimensional structural and electronic properties [2]. Unlike classical 2D-QSAR methods that utilize molecular descriptors such as logP or molar refractivity, 3D-QSAR techniques employ spatial molecular field properties to establish predictive models that can guide molecular optimization [102]. These methods have become indispensable tools for medicinal chemists seeking to understand the structural basis of biological activity, particularly when the three-dimensional structure of the target protein remains unknown [2]. The successful application and reporting of 3D-QSAR studies, however, demand strict adherence to established computational protocols and validation standards to ensure the generation of robust, predictive models that can reliably inform drug design campaigns.
Core Methodologies and Comparative Analysis
Alignment-Dependent Methods
Comparative Molecular Field Analysis (CoMFA) stands as the pioneering 3D-QSAR technique that established the molecular field analysis paradigm [102]. The methodology involves placing aligned molecules within a 3D grid and calculating steric (Lennard-Jones) and electrostatic (Coulombic) interaction energies between a probe atom and the molecules at each grid point [2] [102]. These interaction energies serve as descriptors that are correlated with biological activity using Partial Least Squares (PLS) regression. The performance of CoMFA models is highly dependent on several critical factors, including the quality of biological data, accuracy of molecular alignment, grid resolution, and probe selection [102].
Comparative Molecular Similarity Indices Analysis (CoMSIA) extends beyond CoMFA by incorporating additional molecular fields including hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields [46] [7]. A distinct advantage of CoMSIA lies in its use of a Gaussian function to calculate molecular similarity indices, which eliminates the abrupt energy changes encountered in CoMFA and often produces more interpretable contour maps [7]. Recent studies applying CoMSIA to anti-breast cancer agents and mIDH1 inhibitors have demonstrated its continued relevance, with models exhibiting high correlation coefficients (R² > 0.99) and significant predictive power (Q² > 0.77) [46] [7].
Alignment-Independent Methods
Alignment-independent 3D-QSAR approaches address one of the most significant challenges in conventional 3D-QSAR: the requirement for correct molecular superposition. Techniques such as Grid-INdependent Descriptors (GRIND) utilize molecular interaction fields but derive alignment-independent descriptors by capturing the most relevant product pairs between different field types [103]. Similarly, 3D-Spectral Data-Activity Relationship (3D-SDAR) employs NMR chemical shifts and interatomic distances to generate unique molecular fingerprints without requiring alignment [48]. Studies on androgen receptor binders have demonstrated that these alignment-independent methods can achieve predictive accuracy comparable to alignment-dependent approaches while significantly reducing computational overhead and subjectivity [48].
Table 1: Comparison of Major 3D-QSAR Techniques
| Method | Descriptor Basis | Molecular Fields | Alignment Requirement | Key Advantages |
|---|---|---|---|---|
| CoMFA [102] | Steric & electrostatic energy values at grid points | Steric, Electrostatic | Yes | Established method with straightforward interpretation |
| CoMSIA [46] [7] | Similarity indices using Gaussian function | Steric, Electrostatic, Hydrophobic, H-bond Donor, H-bond Acceptor | Yes | Additional fields; smoother potential functions |
| GRIND [103] | Correlograms of MIF product pairs | Multiple MIF types | No | Alignment-independent; captures crucial long-distance interactions |
| 3D-SDAR [48] | NMR chemical shifts & interatomic distances | Electronic environment | No | Uses experimental NMR data; alignment-independent |
| Pharmacophore Modeling [9] [104] | Spatial arrangement of chemical features | HBA, HBD, Hydrophobic, Aromatic, etc. | Yes (for some approaches) | Direct identification of critical interaction features |
Experimental Protocols and Workflow
Data Set Preparation and Molecular Modeling
The foundation of any robust 3D-QSAR model lies in the careful preparation of the dataset and molecular structures. The following protocol outlines the critical steps:
Data Curation: Compile biological activity data (preferably IC₅₀, EC₅₀, or Kᵢ values) measured under consistent experimental conditions [102]. Convert activity values to pIC₅₀ or pEC₅₀ (-logIC₅₀ or -logEC₅₀) to ensure normal distribution for modeling [9] [103]. A sufficient number of compounds is crucial, with recent studies utilizing datasets ranging from 47 to 62 compounds [9] [103] [7].
Training/Test Set Division: Randomly divide the dataset into training (typically 75-85%) and test (15-25%) sets, ensuring both sets span the entire activity range and contain structurally diverse compounds [9] [104] [7].
Molecular Structure Generation and Optimization: Generate 3D molecular structures from 2D representations using builder panels in molecular modeling software. Conduct geometry optimization using force fields such as MM+ or OPLS_2005, followed by further refinement with semi-empirical methods (e.g., AM1) or density functional theory (DFT) calculations [103] [104].
Conformational Analysis and Bioactive Conformation Selection: Generate multiple low-energy conformers for each compound (typically within 20 kcal/mol of the global minimum) using poling algorithms or systematic searches [104] [105]. Select putative bioactive conformations using methods such as:
Diagram 1: 3D-QSAR Model Development Workflow
Model Development and Validation
The development and validation of 3D-QSAR models require meticulous statistical analysis and rigorous validation protocols:
Molecular Alignment: Align training set molecules according to their putative bioactive conformations using atom-based fitting, pharmacophore feature alignment, or field-based alignment methods [9] [7].
Field Calculation and PLS Analysis: Calculate molecular interaction fields (steric, electrostatic, hydrophobic, etc.) using appropriate probes at grid points surrounding the aligned molecules. Construct the descriptor matrix and correlate with biological activity using Partial Least Squares (PLS) regression to derive the 3D-QSAR model [102] [7].
Model Validation: Employ multiple validation strategies to assess model robustness and predictive power:
- Internal Validation: Calculate cross-validated correlation coefficient (Q²) using leave-one-out or leave-group-out methods. Q² > 0.5 is generally considered acceptable, with Q² > 0.7 indicating excellent predictive ability [9] [7].
- External Validation: Predict activities of test set compounds not included in model building. Calculate predictive R² (R²pred) with values > 0.6 indicating good external predictive ability [46] [7].
- Randomization Tests: Perform Y-randomization to ensure model validity (correlation should be lost when activities are randomly shuffled) [9] [104].
- Statistical Parameters: Report conventional correlation coefficient (R²), standard error of estimate (SEE), F-test value, and optimal number of principal components for complete statistical characterization [9] [7].
Table 2: Essential Statistical Metrics for 3D-QSAR Model Validation
| Validation Type | Statistical Metric | Acceptance Criteria | Interpretation |
|---|---|---|---|
| Internal Validation | Q² (Cross-validated R²) | Q² > 0.5 (Good), Q² > 0.7 (Excellent) | Measure of model predictive ability based on training set |
| External Validation | R²pred (Predictive R²) | R²pred > 0.6 | Measure of model performance on unseen test set compounds |
| Goodness-of-Fit | R² (Correlation coefficient) | R² > 0.8, Close to 1.0 | Measure of how well model fits the training data |
| Goodness-of-Fit | SEE (Standard Error of Estimate) | As low as possible | Measure of precision of the model |
| Model Significance | F-value | Higher values preferred | Measure of statistical significance of the model |
| Randomization Test | Y-Randomization correlation | Significant degradation from original model | Confirms model not based on chance correlation |
Best Practices for Reporting 3D-QSAR Studies
Comprehensive reporting of 3D-QSAR studies is essential for scientific reproducibility and meaningful application of results. The following elements should be explicitly documented:
Data Source and Preparation: Clearly describe the source of biological activity data, measurement conditions, and any data transformation methods (e.g., pIC₅₀ conversion). Specify the rationale for training/test set division and provide structures of all compounds with their experimental and predicted activities [9] [104].
Computational Methods Detail: Document software packages and versions used, molecular optimization protocols (force fields, convergence criteria), conformational search methods (algorithm, energy window, maximum conformers), and alignment strategies (method, template molecule) [9] [103] [104].
Model Building Parameters: Report grid type and dimensions, probe atoms used, field types calculated, PLS parameters, and variable selection methods (if applicable) [102] [7].
Complete Statistical Reporting: Present all relevant statistical parameters including R², Q², SEE, F-value, number of optimal components, and validation results. Include scatter plots of predicted vs. experimental activities for both training and test sets [9] [7].
Contour Map Interpretation: Provide detailed interpretation of 3D contour maps in the context of molecular structure and activity, highlighting regions where specific molecular features enhance or diminish biological activity [9] [46].
Experimental Application: Describe how model insights were translated into molecular design, including synthesis of new compounds, their predicted and experimental activities, and correlation with model predictions [100] [46] [105].
Table 3: Key Research Reagent Solutions for 3D-QSAR Studies
| Resource Category | Specific Tools/Software | Function in 3D-QSAR Workflow |
|---|---|---|
| Molecular Modeling Suites | Schrodinger Suite [9], Accelrys Discovery Studio [104], HyperChem [103] | Provides integrated environment for molecular structure building, optimization, conformational analysis, and QSAR model development |
| 3D-QSAR Specific Software | SYBYL (CoMFA, CoMSIA) [46] [7], GRID [2], ALMOND (GRIND) [103] | Generates molecular interaction fields and alignment-independent descriptors for 3D-QSAR model construction |
| Conformational Analysis Tools | MacroModel [105], CONFORT, Omega | Performs systematic conformational searching and analysis to identify bioactive conformations |
| Docking Software | AutoDock, GOLD, Glide [9] | Determines putative binding modes when protein structure is available to guide molecular alignment |
| Chemical Databases | PubChem [103], IBScreen [9], Zinc | Sources of chemical structures for virtual screening and test set compounds |
| Statistical Analysis | R, MATLAB, PLS toolkits | Performs partial least squares regression and statistical validation of QSAR models |
The rigorous application and comprehensive reporting of 3D-QSAR studies following established best practices enables the development of robust, predictive models that significantly accelerate drug discovery efforts. By carefully addressing each step of the workflow—from data curation and molecular modeling to statistical validation and contour map interpretation—researchers can extract meaningful structure-activity insights that reliably guide molecular design. The integration of 3D-QSAR with complementary computational approaches such as molecular docking and molecular dynamics simulations further enhances the utility of these models in rational drug design. As the field advances, adherence to these protocols will ensure the continued productivity of 3D-QSAR as a valuable tool in the medicinal chemist's arsenal.
Conclusion
CoMFA and CoMSIA represent powerful, well-established methodologies in the computational drug discovery toolkit, providing critical insights into the structural determinants of biological activity. When properly implemented with rigorous statistical validation, these 3D-QSAR techniques offer tremendous value for lead optimization and the rational design of novel therapeutic agents. The future of 3D-QSAR lies in its integration with advanced machine learning approaches, such as CNN-based models that show improved predictive power, and its synergistic application with molecular dynamics simulations and structure-based design. As these methodologies continue to evolve, they will play an increasingly vital role in addressing complex challenges in biomedical research, particularly in the design of multi-target ligands for complex diseases and overcoming drug resistance mechanisms, ultimately accelerating the development of more effective and selective therapeutics.
References
- 1. Three Dimensional Quantitative Structure Activity ... [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/three-dimensional-quantitative-structure-activity-relationship]
- 2. 3D-QSAR - Drug Design Org [https://drugdesign.org/chapters/3d-qsar/]
- 3. Recent Developments in 3D QSAR and Molecular Docking ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7123761/]
- 4. 3.2. CoMFA and CoMSIA Models [https://bio-protocol.org/exchange/minidetail?id=6533469&type=30]
- 5. Machine learning-based 3D-QSAR models for predicting ... [https://www.sciencedirect.com/science/article/pii/S3050787125000137]
- 6. Science Brief: 3D-QSAR Machine Learning for Binding ... [https://www.eyesopen.com/science-briefs/3d-qsar-machine-learning]
- 7. 3D-QSAR, Scaffold Hopping, Virtual Screening, and ... [https://www.mdpi.com/1422-0067/25/13/7434]
- 8. 3D-QSAR, docking, molecular dynamics simulation and ... [https://www.sciencedirect.com/science/article/pii/S1878535217301788]
- 9. 3D-QSAR-Based Pharmacophore Modeling, Virtual ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8410400/]
- 10. 3D-QSAR Design of New Bcr-Abl Inhibitors Based on Purine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12195648/]
- 11. Beginner's Guide to 3D-QSAR in Drug Design [https://neovarsity.org/blogs/beginners-guide-3d-qsar-drug-design]
- 12. Comparative Molecular Field Analysis (CoMFA) and ... [https://www.mdpi.com/1422-0067/12/10/7022]
- 13. Comparative Molecular Similarity Indices Analysis [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/comparative-molecular-similarity-indices-analysis]
- 14. Comparative Molecular Similarity Indices Analysis [https://www.sciencedirect.com/topics/immunology-and-microbiology/comparative-molecular-similarity-indices-analysis]
- 15. Comparative molecular similarity index analysis (CoMSIA ... [https://pubmed.ncbi.nlm.nih.gov/10087495/]
- 16. CoMFA and CoMSIA analysis [https://bio-protocol.org/exchange/minidetail?id=2515250&type=30]
- 17. 3D QSAR pharmacophore, CoMFA and CoMSIA based ... [https://pubmed.ncbi.nlm.nih.gov/22741782/]
- 18. Molecular Fingerprint-based Artificial Neural Networks QSAR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3462244/]
- 19. Py-CoMSIA: An Open-Source Implementation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11945924/]
- 20. CoMFA, CoMSIA, HQSAR and Molecular Docking Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4852576/]
- 21. Structure-Activity Relationship Studies Based on 3D-QSAR ... [https://www.mdpi.com/1420-3049/27/22/7974]
- 22. Three-dimensional quantitative structure–activity ... [https://www.sciencedirect.com/science/article/pii/S0045653503005745]
- 23. Structure-Activity Relationships Based on 3D-QSAR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6099677/]
- 24. Application of 3D QSAR CoMFA/CoMSIA and in silico ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523409001974]
- 25. 2.3. CoMFA and CoMSIA setup [https://bio-protocol.org/exchange/minidetail?id=7214199&type=30]
- 26. 3D-QSAR, molecular docking and molecular dynamics ... [https://www.sciencedirect.com/science/article/pii/S2405844022032790]
- 27. The Top 10 Software for QSAR Analysis [https://parssilico.com/blogs/78-top-10-software-for-qsar-analysis]
- 28. Top Drug Discovery Software Solutions to Watch in 2025 [https://www.deepmirror.ai/post/top-drug-discovery-software-solutions-to-watch-in-2025]
- 29. A Comparative Analysis of Cheminformatics Platforms [https://intuitionlabs.ai/articles/cheminformatics-platforms-comparison]
- 30. Machine Learning 101: How to train your first QSAR model [https://optibrium.com/knowledge-base/machine-learning-101-how-to-train-your-first-qsar-model-blog/]
- 31. QSAR Toolbox [https://qsartoolbox.org/]
- 32. Combined 3D-QSAR modeling and molecular docking ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4531791/]
- 33. Comparative Molecular Field Analysis - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/comparative-molecular-field-analysis]
- 34. Ten quick tips to perform meaningful and reproducible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12064015/]
- 35. Template-Based Method for Conformation Generation and Scoring for Congeneric Series of Ligands - PubMed [https://pubmed.ncbi.nlm.nih.gov/31045363/]
- 36. SkeleDock: Exploiting structural knowledge [TUTORIAL] [https://medium.com/playmolecule/skeledock-exploiting-structural-knowledge-tutorial-37ea3dfc03de]
- 37. MolCompass: multi-tool for the navigation in chemical space ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11318166/]
- 38. Combined 3D-QSAR, molecular docking and dynamics ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.1003816/full]
- 39. Design, 3D-QSAR, molecular docking, MD simulations ... [https://www.sciencedirect.com/science/article/abs/pii/S0022286025000407]
- 40. Quinoxaline derivatives in identifying novel VEGFR-2 ... [https://pubmed.ncbi.nlm.nih.gov/40850176/]
- 41. 3D-QSAR and Molecular docking of Indolo[3,2-c] ... [https://theaspd.com/index.php/ijes/article/view/6021]
- 42. Protocol for Molecular Dynamics Simulations of Proteins [https://en.bio-protocol.org/en/bpdetail?id=2051&type=0]
- 43. Introduction - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/2025.3/reference-manual/introduction.html]
- 44. Schrödinger Release Notes - Release 2025-3 [https://www.schrodinger.com/life-science/download/release-notes/release-2025-3/]
- 45. The three secrets to great 3D-QSAR: Alignment, alignment ... [https://cresset-group.com/about/news/the-three-secrets-to-great-3d-qsar-alignment-align/]
- 46. Design, 3D-QSAR, molecular docking, ADMET ... [https://www.sciencedirect.com/science/article/pii/S2667022423002943]
- 47. 3D-QSAR Studies, Molecular Docking, Molecular Dynamic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9865323/]
- 48. Alignment-independent technique for 3D QSAR analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4833814/]
- 49. Pentacle - Advanced Alignment-Independent 3D QSAR [https://www.moldiscovery.com/software/pentacle/]
- 50. Automatic generation of alignments for 3D QSAR analyses [https://www.sciencedirect.com/science/article/abs/pii/S1093326301001103]
- 51. PharmQSAR - 3D QSAR Software Package [https://pharmacelera.com/pharmqsar/]
- 52. Exhaustive CoMFA and CoMSIA analyses around different ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6009877/]
- 53. 3D-QSAR - Lead Optimization in Orion [https://www.eyesopen.com/3d-qsar]
- 54. A study of hydrophobic domain formation of polymeric drug ... [https://www.sciencedirect.com/science/article/pii/S0928098724001039]
- 55. Water-Based Pharmacophore Modeling in Kinase Inhibitor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12458683/]
- 56. Tutorial: Molecular Descriptors in QSAR [https://people.chem.ucsb.edu/kahn/kalju/chem162/public/molecules_qsar.html]
- 57. Comparative molecular moment analysis (CoMMA): 3D ... [https://pubmed.ncbi.nlm.nih.gov/8667357/]
- 58. Molecular Descriptors - Computational Chemistry Glossary [https://deeporigin.com/glossary/molecular-descriptors]
- 59. Water Pharmacophore: Designing Ligands using ... [https://www.nature.com/articles/s41598-018-28546-z]
- 60. 3D-QSAR CoMFA study of some Heteroarylpyrroles as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2792495/]
- 61. Design, Quantitative Structure–Activity Relationships and ... [https://www.sciencedirect.com/science/article/pii/S2667022425001458]
- 62. Comparative Molecular Field Analysis - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/comparative-molecular-field-analysis]
- 63. Application of 3D-QSAR, Pharmacophore, and Molecular ... [https://www.mdpi.com/1422-0067/19/5/1436]
- 64. 3D-QSAR and molecular docking studies on HIV protease ... [https://www.sciencedirect.com/science/article/abs/pii/S0022286016309784]
- 65. A Case Study on HIV-1 Protease Inhibitors [https://pubmed.ncbi.nlm.nih.gov/21547550/]
- 66. Towards designing of a potential new HIV-1 protease ... [https://pubmed.ncbi.nlm.nih.gov/37079533/]
- 67. CoMFA, CoMSIA and Topomer CoMFA descriptors [https://bio-protocol.org/exchange/minidetail?id=3616167&type=30]
- 68. Computational studies on HIV-1 protease inhibitors [https://pubmed.ncbi.nlm.nih.gov/11087569/]
- 69. Py-CoMSIA: An Open-Source Implementation of ... [https://www.mdpi.com/1424-8247/18/3/440]
- 70. 3D-QSAR (CoMFA/CoMSIA) & Alignment Strategies [https://www.nthrys.com/bioinformatics-training/3d-qsar-comfa-comsia-and-alignment-strategies.html]
- 71. Design and evaluation of novel triazole derivatives as ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1518777/full]
- 72. Integration of machine learning in 3D-QSAR CoMSIA models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10654693/]
- 73. 2.3. Generation of 3D-QSAR models [https://bio-protocol.org/exchange/minidetail?id=5980533&type=30]
- 74. Integration of machine learning in 3D-QSAR CoMSIA ... [https://pubs.rsc.org/en/content/articlelanding/2023/ra/d3ra06690h]
- 75. A Beginner's Guide to QSAR Modeling in Cheminformatics ... [https://neovarsity.org/blogs/cheminformatics-qsar-beginner-guides]
- 76. 3D-QSAR (CoMFA, CoMSIA), molecular docking and ... [https://www.sciencedirect.com/science/article/abs/pii/S0960894X17305632]
- 77. A Record-Breaking Dataset to Train AI Models has Launched [https://newscenter.lbl.gov/2025/05/14/computational-chemistry-unlocked-a-record-breaking-dataset-to-train-ai-models-has-launched/]
- 78. Improvement of the Prediction Power of the CoMFA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3447613/]
- 79. Combined 3D-QSAR, molecular docking and dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9666879/]
- 80. 3D-QSAR model development [https://bio-protocol.org/exchange/minidetail?id=3309422&type=30]
- 81. Docking and scoring - Schrödinger [https://www.schrodinger.com/life-science/learn/white-papers/docking-and-scoring/]
- 82. AutoGPA: An Automated 3D-QSAR Method Based on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4207448/]
- 83. Two Decades of 4D-QSAR: A Dying Art or Staging ... [https://www.mdpi.com/1422-0067/22/10/5212]
- 84. 3D-QSAR, design, molecular docking and dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11841475/]
- 85. 3D-QSAR, design, molecular docking and dynamics ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1545791/full]
- 86. The importance of choosing a proper validation strategy in ... [https://www.sciencedirect.com/science/article/pii/S0003267023007535]
- 87. Cross-validation (statistics) [https://en.wikipedia.org/wiki/Cross-validation_(statistics)]
- 88. 3.1. Cross-validation: evaluating estimator performance [https://scikit-learn.org/stable/modules/cross_validation.html]
- 89. Practical Considerations and Applied Examples of Cross ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11041453/]
- 90. Reliable estimation of prediction errors for QSAR models ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-014-0047-1]
- 91. Modelling methods and cross-validation variants in QSAR [https://pubmed.ncbi.nlm.nih.gov/30160175/]
- 92. 3D-QSAR AND CONTOUR MAP ANALYSIS OF ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4762489/]
- 93. Rational design of novel phenol ether derivatives as non ... [https://www.explorationpub.com/Journals/eds/Article/100829]
- 94. 3D-QSAR CoMFA/CoMSIA models based on theoretical ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523409003080]
- 95. Comparison to 3rd-party Software — Orion Workflows 2025.2.2 ... [https://docs.eyesopen.com/floe/modules/models_3dqsar/docs/source/benchmarks/results.html]
- 96. Comparison of various methods for validity evaluation of ... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-022-00856-4]
- 97. AI-Integrated QSAR Modeling for Enhanced Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12525248/]
- 98. An improved 3D quantitative structure-activity relationships ... [https://www.sciencedirect.com/science/article/pii/S2667318523000090]
- 99. Development of convolutional neural network-based QSAR ... [https://jksus.org/development-of-convolutional-neural-network-based-qsar-model-to-predict-cardiotoxicity-and-principal-component-analysis-fingerprint-analysis/]
- 100. Application of CoMFA and CoMSIA 3D-QSAR and docking ... [https://pubmed.ncbi.nlm.nih.gov/14711310/]
- 101. Advancing QSAR models in drug discovery for best ... [https://www.sciencedirect.com/science/article/abs/pii/S0169743925002291]
- 102. Introduction to 3D-QSAR [https://basicmedicalkey.com/introduction-to-3d-qsar/]
- 103. Alignment independent 3D-QSAR studies and molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7110456/]
- 104. Development, evaluation and application of 3D QSAR ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-S14-S4]
- 105. 3D-QSAR studies and synthesis of novel antiplasmodial ... [https://www.sciencedirect.com/science/article/abs/pii/S0960894X15300421]