Beyond Maximum Tolerated Dose: A Mathematical Modeling Approach to Optimizing Combination Therapy in Oncology
The paradigm for dosing oncology drugs is shifting from the traditional maximum tolerated dose (MTD) approach toward model-informed, optimized strategies, particularly for modern targeted therapies and immunotherapies.
Beyond Maximum Tolerated Dose: A Mathematical Modeling Approach to Optimizing Combination Therapy in Oncology
Abstract
The paradigm for dosing oncology drugs is shifting from the traditional maximum tolerated dose (MTD) approach toward model-informed, optimized strategies, particularly for modern targeted therapies and immunotherapies. This article explores the transformative role of mathematical modeling in optimizing combination therapy doses, a critical challenge in cancer treatment. We cover the foundational principles of mathematical oncology, detail key methodological approaches like Quantitative Systems Pharmacology and exposure-response modeling, and examine their application in clinical trial design. The article also addresses troubleshooting for common hurdles like stromal-induced resistance and outlines validation frameworks through recent clinical trials and regulatory initiatives like Project Optimus. Aimed at researchers, scientists, and drug development professionals, this review synthesizes how computational models are paving the way for more effective, personalized, and less toxic combination cancer therapies.
The New Paradigm: Why Mathematical Models are Replacing Maximum Tolerated Dose
The Limitation of the MTD Paradigm for Modern Therapies
Troubleshooting Guide: Common Challenges in Moving Beyond MTD
Problem 1: High rates of dose reduction in late-stage trials.
- Root Cause: Reliance on the traditional 3+3 dose-escalation design, which focuses on short-term dose-limiting toxicities (DLTs) and fails to represent the long treatment courses of modern therapies [1].
- Solution: Implement novel trial designs like model-informed dose escalation and randomized dose-selection sub-studies to better characterize the dose-response relationship and long-term tolerability [2] [1].
Problem 2: Rapid emergence of drug resistance and treatment failure.
- Root Cause: The MTD approach applies intense evolutionary pressure, eliminating drug-sensitive cells and causing "competitive release" of resistant populations [3] [4].
- Solution: Utilize adaptive therapy strategies, informed by mathematical models (e.g., Lotka-Volterra systems), to maintain sensitive cells and suppress resistant growth [3] [5].
Problem 3: Inefficient dose optimization delaying drug development.
- Root Cause: Attempting to optimize dosage only in post-approval settings, which can take years [6].
- Solution: Integrate dose optimization early in preclinical development using quantitative systems pharmacology models and exposure-response analyses [2] [1].
Frequently Asked Questions (FAQs)
FAQ 1: Why is the traditional Maximum Tolerated Dose (MTD) paradigm no longer suitable for many modern cancer drugs? The MTD paradigm, developed for cytotoxic chemotherapies, is based on determining the highest dose patients can tolerate in a short first course of treatment [2]. This approach is suboptimal for targeted therapies and immunotherapies because their mechanism of action differs; efficacy often saturates at a certain level, and higher doses only increase toxicity without improving efficacy [5]. Studies show that nearly 50% of patients on targeted therapies require dose reductions, and the FDA has mandated post-approval dose re-evaluation for over 50% of recently approved cancer drugs [1].
FAQ 2: What is the alternative to the MTD approach? The leading alternative is dose optimization, which aims to identify the Optimal Biological Dose (OBD) that best balances efficacy and tolerability [2]. This involves:
- Using randomized trials to compare multiple doses before approval [6].
- Employing model-informed drug development (MIDD) and pharmacokinetic/pharmacodynamic (PK/PD) modeling [1].
- Shifting towards adaptive therapy strategies that control tumor growth rather than seeking maximal cell kill [3] [4].
FAQ 3: How can mathematical modeling improve dose selection for combination therapies? Mathematical models, such as Lotka-Volterra competition models, help simulate complex eco-evolutionary dynamics within tumors during treatment [3] [5]. For combination therapy, they can:
- Predict the emergence of resistance to multiple drugs.
- Identify synergistic dosing schedules that exploit fitness costs to resistant subpopulations.
- Optimize "first-strike, second-strike" sequential therapy strategies to delay or prevent resistance [5] [4].
FAQ 4: What trial designs are recommended for dose optimization? Regulatory guidance now encourages randomized dose-finding trials before approval [2]. Recommended designs include:
- Model-informed designs: Utilize mathematical modeling for more nuanced dose-escalation/de-escalation based on efficacy and late-onset toxicities [1].
- Adaptive seamless designs: Combine traditionally separate trial phases (e.g., FIH and proof-of-concept) to accelerate enrollment and gather more long-term data [1].
- Backfill and expansion cohorts: Increase patient numbers at specific dose levels within early-stage trials to strengthen understanding of the benefit/risk ratio [1].
FAQ 5: What data should be collected to inform the Optimal Biological Dose (OBD)? Dose selection should be justified by a totality of evidence, moving beyond just early-cycle toxicities [2]. Key data includes:
- Longitudinal patient-reported outcomes (PROs) and quality-of-life metrics [2].
- Pharmacokinetic/Pharmacodynamic (PK/PD) and exposure-response relationships [1].
- Biomarker data (e.g., ctDNA dynamics) to identify early response signals [1].
- Cumulative and delayed toxicities occurring after cycle 1 [2].
Quantitative Data on MTD Limitations and New Paradigms
Table 1: Documented Limitations of the MTD Paradigm
| Metric | Finding | Source |
|---|---|---|
| Patient Dose Reductions | Nearly 50% of patients on late-stage trials of small molecule targeted therapies required dose reductions due to side effects. | [1] |
| FDA Post-Approval Actions | Over 50% of recently approved cancer drugs required additional studies to re-evaluate dosing. | [1] |
| Patient-Reported Toxicity | 86% of patients with metastatic breast cancer reported significant treatment-related side effects. | [6] |
| Clinician Support for Change | Over 80% of surveyed oncologists strongly supported future trials focused on optimal dose determination over MTD. | [6] |
Table 2: Key Mathematical Models for Therapy Optimization
| Model Type | Primary Application | Key Function |
|---|---|---|
| Lotka-Volterra Competition Models | Adaptive Therapy | Models competition between drug-sensitive and resistant cell populations to design therapy schedules that suppress resistance [3]. |
| Pharmacokinetic-Pharmacodynamic (PK/PD) Models | Dose-Response Characterization | Links drug exposure (pharmacokinetics) to biological effect (pharmacodynamics) to predict efficacy and toxicity [1] [5]. |
| Quantitative Systems Pharmacology (QSP) | Final Dosage Decision | Integrates larger clinical datasets to identify optimized dosages, extrapolate effects of untested schedules, and address confounders [1]. |
| Bang-Bang Control Theory | Intermittent vs. Continuous Dosing | Formally analyzes intermittent adaptive therapy and proves robustness of continuous adaptive therapy [3]. |
Experimental Protocols for Dose Optimization Research
Protocol 1: Implementing a Model-Informed First-in-Human (FIH) Trial
Objective: To identify a range of safe and potentially effective doses for further study, moving beyond the algorithmic 3+3 design [1].
Methodology:
- Starting Dose Selection: Use mathematical models that go beyond animal weight-based scaling. Incorporate factors like receptor occupancy differences between species to determine a higher, potentially more efficacious, starting dose [1].
- Dose Escalation: Employ model-informed dose escalation designs (e.g., continual reassessment method). These designs use statistical models to continuously update the probability of toxicity and efficacy based on all accumulated data from previous patients, allowing for more efficient and nuanced dose escalation/de-escalation decisions [1].
- Data Collection: Collect pharmacokinetic (PK) samples, early pharmacodynamic (PD) biomarkers (e.g., ctDNA), and monitor for both early and late-onset toxicities [1].
Protocol 2: Randomized Dose-Selection (Proof-of-Concept) Study
Objective: To directly compare multiple doses and identify the leading candidate for registrational trials [2] [1].
Methodology:
- Dose Selection: Choose 2-3 doses from the FIH trial that span a range of biological activity and safety profiles.
- Trial Design: Randomize patients to the different dose arms. Incorporate backfill or expansion cohorts to enroll more patients at doses of particular interest [1].
- Endpoint Assessment: Assess antitumor activity (e.g., objective response rate), safety, tolerability, and patient-reported outcomes (PROs). Integrate biomarker data (e.g., ctDNA) for early response detection [1].
- Decision Framework: Use a quantitative framework like a Clinical Utility Index (CUI) to integrate all available efficacy, safety, and PK/PD data and justify the final dose selection [1].
Protocol 3: Calibrating a Mathematical Model for Adaptive Therapy
Objective: To develop a patient-specific model for predicting response to adaptive dosing schedules [3] [5].
Methodology:
- Model Selection: Use a two-population Lotka-Volterra system to represent sensitive (x) and resistant (y) cell dynamics:
dx/dt = r_x * x * (1 - x - α * y) - K_A(x,y,t) * h(x, r_d)dy/dt = r_y * y * (1 - y - β * x)whereris growth rate,αandβare competition coefficients, andK_Ais the treatment function [3]. - Parameter Estimation: Calibrate model parameters (rx, ry, α, β) using longitudinal tumor burden data (e.g., from imaging or PSA levels) from historical cohorts or the patient's initial treatment cycles [5].
- Schedule Simulation: Simulate different therapy schedules (continuous fixed-dose, intermittent on-off, continuous adaptive) to predict which strategy maximizes time to progression for that specific patient [3].
- Validation: Compare model predictions to actual patient outcomes in pilot clinical trials (e.g., NCT03543969 for melanoma, NCT02415621 for prostate cancer) [5].
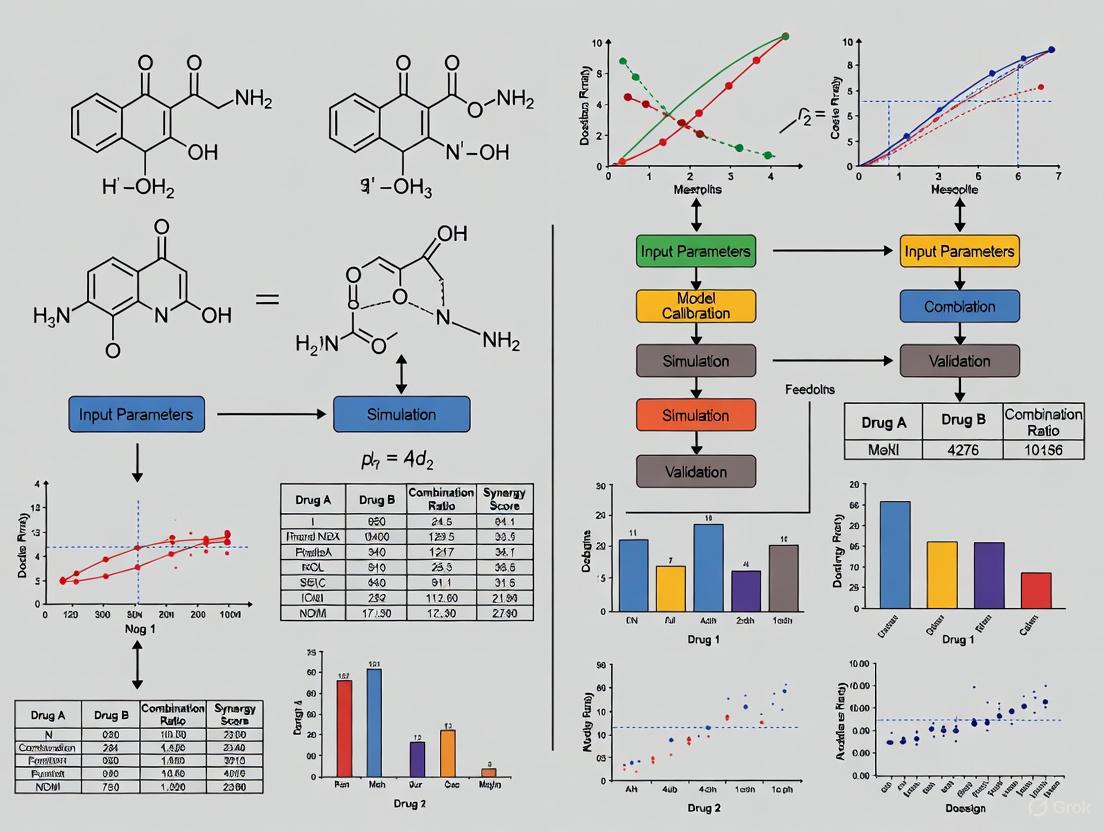
Diagram: Workflow for Model-Informed Dose Optimization
Model-Informed Dose Optimization Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Dose Optimization Research
| Tool / Reagent | Function in Research |
|---|---|
| Circulating Tumor DNA (ctDNA) | A liquid biopsy biomarker used to track tumor burden and response dynamics early in treatment, informing dose-response relationships [1]. |
| Patient-Reported Outcome (PRO) Measures | Standardized questionnaires to capture the patient's perspective on treatment side effects and quality of life, critical for evaluating the tolerability of different doses [2]. |
| Lotka-Volterra Competition Model | A system of differential equations used to model the competitive interaction between drug-sensitive and drug-resistant cancer cell populations under treatment pressure [3]. |
| Clinical Utility Index (CUI) | A quantitative framework that integrates multiple endpoints (efficacy, toxicity, PROs) into a single score to aid in collaborative and objective dose selection [1]. |
| Population PK/PD Models | Mathematical models that quantify the relationship between drug dose, systemic exposure (pharmacokinetics), and biological effect (pharmacodynamics) across a patient population [1]. |
| S1P1 Agonist III | S1P1 Agonist III, MF:C21H16F3N3O3, MW:415.4 g/mol |
| TCH-165 | TCH-165, MF:C39H37N3O3, MW:595.7 g/mol |
Frequently Asked Questions (FAQs)
Q1: What is the primary clinical challenge when combining multiple anti-cancer drugs, and how can mathematical oncology help? A key challenge is determining safe and effective starting doses for novel drug combinations, especially those involving both targeted and cytotoxic agents. Mathematical modeling analyzes historical clinical trial data to establish that for three-drug combinations, less than 30% of studies could administer all three drugs at their full single-agent dose. Dose reductions to as low as 45% of each single agent's dose are often required. Modeling provides a quantitative framework to predict safe additive dose percentages, helping to avoid excessive toxicity in early-phase trials [7].
Q2: My mathematical model fits the training data well but fails to predict unseen test data. What should I do? This is a common step in the modeling workflow. A model that fails to predict unseen data is not necessarily "wrong"; it often indicates that the model lacks a biological process that becomes important under the new conditions (e.g., in vivo versus in vitro). Use this failure to refine your model. For instance, if the model accurately predicts the first 8 days of tumor spheroid growth but fails thereafter, consider if processes like drug resistance, immune responses, or angiogenesis—not accounted for in the original model—begin to dominate at that point. This failure provides a critical opportunity to challenge the model's assumptions and integrate new biology [8].
Q3: How can optimal control theory be applied to combination therapy? Optimal control theory can determine dosage protocols that steer a patient's state from a malignant condition (tumor escape) to a benign one (equilibrium). This involves formulating a mathematical model of tumor-immune-drug interactions and then solving for the time-varying dose rates (controls) that minimize an objective function, which typically balances tumor burden and drug toxicity. The solution suggests how to schedule chemo- and immunotherapy over time to leverage their synergistic effects, such as the immune-stimulatory release of tumor antigens following chemotherapy [9].
Q4: What are the "Three E's of Immuno-editing" in mathematical models? This qualitative framework describes the possible long-term outcomes of tumor-immune interactions modeled by dynamical systems [9]:
- Elimination: The immune system eradicates the tumor. The tumor-free stationary point is stable.
- Equilibrium: The immune system controls the tumor, maintaining it in a dormant state. This is represented by a stable stationary point with a low, positive tumor volume.
- Escape: The tumor grows uncontrollably as it evades the immune response. This corresponds to an unstable stationary point with a high tumor volume. The goal of therapy is to revert an "escape" scenario back to "equilibrium."
Q5: What is the advantage of a dual-target inhibitor over combination therapy? While combining a cytotoxic drug with an epigenetic-targeted drug can overcome the limitations of single-agent therapy, it carries risks of drug-drug interactions, complex pharmacokinetics, and combined toxicity. Dual-target inhibitors are single molecules designed to simultaneously inhibit both an epigenetic and a cytotoxic pathway. This approach can simplify treatment, improve pharmacokinetic profiles, and more effectively overcome compensatory resistance mechanisms that limit single-target drugs [10].
Troubleshooting Guides
Problem: Determining Safe Starting Doses for Novel Drug Combinations
Issue: Designing a first-in-human clinical trial for a new three-drug regimen involving targeted and cytotoxic therapies. The safe starting dose for the combination is unknown, and you wish to avoid excessive toxicity.
Solution: Utilize a model-derived "additive dose percentage" based on historical clinical trial data.
- Background: A systematic review of phase I-III trials provides a benchmark. The "dose percentage" for a drug is defined as (safe dose in combination / single-agent recommended dose) × 100. The "additive dose percentage" is the sum of this value for all three drugs in the combination [7].
- Methodology:
- Identify Combination Type: Classify your planned regimen.
- Consult Reference Data: Use the following table of historical safe additive dose percentages to guide your initial dose selection [7]:
| Combination Type | Number of Studies / Subjects | Median Additive Dose Percentage | Lowest Safe Additive Dose Percentage | Notes |
|---|---|---|---|---|
| 1 Targeted Agent + 2 Cytotoxic Agents | 340 studies / 34,835 subjects | 267% | 137% | Only 28% of studies could give all 3 drugs at 100% dose [7]. |
| 2 Cytotoxic Agents at 100% Dose | 190 studies / 22,454 subjects | 300% | 225% | Applies when the cytotoxic doublet has a known safety profile. Not for HDAC inhibitors [7]. |
| 2 Targeted Agents + 1 Cytotoxic Agent | Information Missing | Information Missing | 133% | Increases to 250% if the two targeted agents are antibodies [7]. |
- Actionable Protocol:
- Calculate the theoretical maximum additive dose percentage (300% if all three drugs can be given at full dose).
- Based on your combination type, select a conservative starting additive dose percentage from the "Lowest Safe" column (e.g., 140-160% for a one-targeted, two-cytotoxic regimen).
- Allocate this total percentage across the three drugs, considering their individual toxicity profiles. For example, you might start with 50% of the targeted agent, 50% of cytotoxic drug A, and 40% of cytotoxic drug B (sum = 140%).
- Use this calculated dose for your initial cohort and proceed with standard phase I dose escalation.
Problem: Designing a Dose-Finding Trial Using Mathematical Modeling
Issue: You need to find the optimal vaccine or drug dose that maximizes efficacy while minimizing toxicity, but testing a large number of doses is impractical.
Solution: Implement a modeling-based dose-optimization approach within your trial design.
- Background: Instead of directly comparing a few doses, this method uses data from all trial participants to fit statistical models that describe the underlying dose-efficacy and dose-toxicity relationships [11].
- Methodology & Protocol:
- Choose Efficacy/Toxicity Models:
- For efficacy, use a binary outcome (e.g., immune response above a threshold). The dose-response can be "saturating" (monotonically increasing) or "peaking" (efficacy decreases after an optimum). Use models like the Latent Quadratic for peaking responses [11]:
Peaking(Dose) = 1 / [1 + e^(base + gradient1 * Dose + gradient2 * Dose^2)] - For toxicity, use an ordinal model (e.g., Grades 0-3) to capture lower-grade events that impact patient quality of life and uptake [11].
- For efficacy, use a binary outcome (e.g., immune response above a threshold). The dose-response can be "saturating" (monotonically increasing) or "peaking" (efficacy decreases after an optimum). Use models like the Latent Quadratic for peaking responses [11]:
- Define Utility Function: Combine the efficacy and toxicity models into a single "utility" function that balances the two, aiming to maximize efficacy and minimize toxicity [11].
- Select Trial Design:
- Fixed-Dose Design: Pre-select doses across the expected range (e.g., low, medium, high).
- Adaptive Design (Recommended): Use continual modeling at interim stages. After each cohort, fit the models to all accumulated data and recommend the dose predicted to have the highest utility for the next cohort. This is more ethical and efficient [11].
- Determine Trial Size: Simulation studies suggest that modeling approaches perform well with at least 30 participants [11].
- Choose Efficacy/Toxicity Models:
Problem: Model Predicts Poor Synergy Between Chemo- and Immunotherapy
Issue: Your model of combination therapy fails to show the synergistic effects observed in some clinical contexts.
Solution: Ensure your model incorporates the immuno-stimulatory effects of cytotoxic drugs.
- Background: Chemotherapy can cause immunogenic cell death, releasing tumor antigens that stimulate an immune response. This is the basis for synergy with immunotherapy [9].
- Troubleshooting Steps:
- Audit Your Model Equations: Check if your model includes a variable for tumor antigen and a term linking its presence to immune cell stimulation.
- Include Key Dynamics: A minimal model should include [9]:
- Tumor Antigen (z):
ż = σx + ψxu - μzσx: Natural antigen production by tumor volume (x).ψxu: Therapy-induced antigen release (a function of tumor volume and drug dose u).μz: Clearance of antigen.
- Immuno-competent Cell Density (y):
Ạ= a(1 - bx)yz + γ - δy - κyu + νyva(1 - bx)yz: Proliferation stimulated by antigen (z).νyv: Boost from immunotherapy (v).
- Tumor Antigen (z):
- Calibrate Parameters: The parameter
ψ(therapy-induced immunogenicity) is critical. If it is set to zero, the synergistic effect will be lost. Consult literature for estimates or calibrate it against data showing the abscopal effect or similar phenomena.
The Scientist's Toolkit: Key Reagent Solutions
| Item / Concept | Function in Mathematical Oncology Research |
|---|---|
| Qualitative Dynamical Systems | Low-dimensional models (ODEs) to understand the totality of possible tumor-immune interactions, such as the "Three E's" of immunoediting. Useful for theoretical insights and optimal control studies [9]. |
| Cell-Based/Agent-Based Models | Computational models that simulate individual cells (agents) in a virtual tissue. Used to explore how single-cell behaviors (e.g., proliferation, death, mutation) lead to emergent tumor-scale dynamics like heterogeneity and drug resistance [12]. |
| Optimal Control Theory | A mathematical framework to compute time-varying dosage protocols (controls) that minimize a cost function (e.g., tumor burden + drug toxicity) subject to the constraints of a dynamical model of cancer treatment [9]. |
| Additive Dose Percentage Metric | A quantitative metric derived from historical clinical trial data to calculate safe starting doses for multi-drug combinations by summing the percentage of each drug's single-agent dose used in the combo [7]. |
| Dose-Utility Function | A function that combines a dose-efficacy model and a dose-toxicity model into a single value. It is maximized to identify the optimal dose that best balances treatment benefit and side effects [11]. |
| Model Averaging | A technique used when the true shape of the dose-response curve is unknown. Predictions from multiple models (e.g., saturating and peaking) are combined, weighted by how well each model fits the data, to make more robust inferences [11]. |
| TCO-amine | TCO-amine, CAS:1609736-43-7, MF:C12H22N2O2, MW:226.32 |
| TCO-C3-PEG3-C3-amine | TCO-C3-PEG3-C3-amine, MF:C19H36N2O5, MW:372.5 g/mol |
Experimental Workflow & Signaling Pathways
Diagram: Optimizing Combination Therapy Doses
Diagram: Three E's of Immuno-Editing & Therapy Goal
Optimizing Combination Therapy Doses Using Mathematical Modeling Research
Troubleshooting Guides
Guide 1: Addressing Poor Dose-Response Model Fitting
Problem: Your dose-response model fails to adequately fit the experimental data, leading to unreliable estimates of potency (e.g., ECâ‚…â‚€) or efficacy (Eₘâ‚â‚“).
Solutions:
- Verify Data Quality: Ensure your experimental design includes an adequate number of data points, especially around the anticipated ECâ‚…â‚€. Confirm that the response measurements are precise and that controls are functioning correctly.
- Re-evaluate Model Selection: The Hill equation is a standard model for sigmoidal dose-response curves [13]. However, if your data does not reach a clear plateau, or if there is a baseline effect, the Emax model, which includes a parameter for the effect at zero dose (Eâ‚€), may be more appropriate [13].
- Check for Non-Monotonicity: In some biological systems, particularly with endocrine disruptors or immune responses, the dose-response relationship may be U-shaped or otherwise non-monotonic [13] [14]. Standard models will not fit this data well. Explore alternative models that can capture this complexity.
- Utilize Appropriate Software: Employ specialized software for dose-response modeling, such as the U.S. EPA's Benchmark Dose Software (BMDS), which provides robust statistical analysis and model averaging techniques [13] [15].
Guide 2: Accounting for Tumor Heterogeneity in Therapy Response
Problem: A combination therapy shows promising results in vitro but fails in vivo or yields highly variable patient responses due to pre-existing or acquired tumor heterogeneity.
Solutions:
- Implement Multiregion Sampling: Do not rely on a single tumor biopsy. Use multiregion sequencing to decode the complex clonal architecture and spatial heterogeneity of the tumor [16].
- Longitudinal Monitoring via Liquid Biopsies: Serial characterization of genetic variants in plasma samples (liquid biopsies) provides a minimally invasive method to track temporal heterogeneity and the emergence of resistant subclones during treatment [16].
- Adopt Combinatorial Strategies: Design therapies that pair agents targeting the dominant, drug-sensitive cell population with agents that target known or predicted resistant subclones. This "vertical" targeting is essential for durable responses [16].
- Incorporate Heterogeneity into Models: When building pharmacokinetic-pharmacodynamic (PK/PD) models, account for the differential sensitivity of distinct tumor subpopulations. This may involve modeling multiple cell types with different response parameters.
Guide 3: Managing Unexpected Eco-Evolutionary Dynamics in Preclinical Models
Problem: During long-term preclinical studies, the system under investigation (e.g., a xenograft model or a microbial infection) evolves, altering the therapy's effectiveness over time.
Solutions:
- Shorten Experiment Duration: If feasible, design studies to be completed within a timeframe that minimizes the opportunity for significant evolutionary change in the target system.
- Model Eco-Evolutionary Feedbacks: Incorporate the potential for rapid evolution into your mathematical models. For example, model predator-prey-like dynamics between the therapy and the tumor cell population, where evolutionary changes in one drive ecological changes in the other [17].
- Design Evolution-Informed Dosing Schedules: Use adaptive therapy principles, where dosing is modulated to maintain a population of therapy-sensitive cells that can outcompete resistant ones, rather than always aiming for maximum cell kill.
Frequently Asked Questions (FAQs)
FAQ 1: What is the fundamental difference between the Hill equation and the Emax model for dose-response analysis?
Both models are used to describe dose-response relationships, but the Emax model is a generalization of the Hill equation. The standard Hill equation assumes the effect starts at zero when the dose is zero [13]. In contrast, the Emax model includes an additional parameter (Eâ‚€) to represent the baseline effect at zero dose, making it more flexible for real-world data where a background effect may be present [13]. The Emax model is considered the most common non-linear model in drug development [13].
FAQ 2: How can mathematical modeling improve dose optimization for oncology combination therapies, as encouraged by Project Optimus?
Project Optimus emphasizes the need for thorough dose optimization rather than simply establishing a maximum tolerated dose [18] [19]. Mathematical modeling is central to this by:
- Integrating Diverse Data: Leveraging non-clinical pharmacology, safety data, and early clinical results to build a holistic picture of the dose-response relationship [19].
- Informing Trial Design: Using model-informed drug development (MIDD) to simulate different dosing schedules and combinations, helping to select the most informative doses for clinical trials and reducing the number of patients exposed to potentially subtherapeutic or overly toxic regimens [19].
- Supporting the "Totality of Evidence": Providing a quantitative framework to justify the selected combination dose to regulators, based on a balance of efficacy and safety [19].
FAQ 3: Why might the infection risk from a total pathogen dose be overestimated if it is administered all at once versus over time?
Traditional dose-response models often assume each pathogen particle carries an independent risk, ignoring immune system dynamics [14]. In reality, the immune system has effectors (e.g., antibodies, macrophages) that can engage and eliminate pathogens. When a dose is spread over time, the immune system has a chance to neutralize earlier arrivals and replenish its effector capacity, reducing the probability that any single pathogen will establish an infection [14]. A model that incorporates these dynamics shows that a dose of 313 Cryptosporidium parvum pathogens given at once had an infection risk of 0.66, but when the same dose was spread over a 100-fold longer window, the risk dropped to 0.09 [14].
FAQ 4: How does tumor heterogeneity drive resistance to combination cancer therapies?
Tumor heterogeneity provides the "fuel for resistance" [16]. A tumor is not a uniform mass of identical cells but a collection of subclones with distinct molecular signatures [16].
- Pre-existing Resistance: Some subclones may harbor intrinsic resistance mutations to one or more drugs in the combination regimen before treatment even begins.
- Therapeutic Selection Pressure: The therapy kills the dominant, drug-sensitive subclones, but inadvertently selects for and allows the expansion of pre-existing resistant minor subclones.
- Acquired Resistance: Under the selective pressure of therapy, some tumor cells may evolve new resistance mechanisms, for instance, through genomic instability [16].
Quantitative Data Tables
Table 1: Key Parameters in Common Dose-Response Models
| Parameter | Definition | Interpretation in Therapy Development |
|---|---|---|
| ECâ‚…â‚€ / ICâ‚…â‚€ | The dose or concentration that produces half of the maximal effect or inhibition. | A measure of potency; a lower value indicates greater potency. |
| Eₘâ‚â‚“ | The maximum achievable effect of the drug. | A measure of efficacy; the theoretical upper limit of the drug's response. |
| Hill Coefficient (n) | Describes the steepness of the dose-response curve. | Reflects cooperativity or the number of molecules binding to a receptor; a steeper curve suggests a narrower therapeutic window. |
| Eâ‚€ | The baseline effect in the absence of the drug. | Accounted for in the Emax model; represents the system's background activity [13]. |
Table 2: Impact of Exposure Dynamics on Infection Risk (Modeling Data)
This table summarizes findings from a model that incorporates immune effector dynamics, demonstrating how the same total dose administered over different time windows leads to different infection risks [14].
| Pathogen | Total Dose | Temporal Exposure Window | Model-Predicted Infection Risk |
|---|---|---|---|
| Cryptosporidium parvum | 313 pathogens | Single, instantaneous dose | 0.66 (66%) |
| Cryptosporidium parvum | 313 pathogens | Spread over 100x longer window | 0.09 (9%) |
Experimental Protocols
Protocol 1: Fitting a Dose-Response Curve to In Vitro Combination Therapy Data
Objective: To quantitatively assess the synergy between two drugs (Drug A and Drug B) using the Hill equation.
Materials:
- Cell line of interest
- Drug A and Drug B stock solutions
- Cell culture plates (96-well)
- Cell viability assay kit (e.g., MTT, CellTiter-Glo)
Methodology:
- Plate Cells: Seed cells at an optimized density in a 96-well plate and allow them to adhere overnight.
- Prepare Drug Dilutions: Create a matrix of serial dilutions for Drug A and Drug B, covering a range from zero effect to maximum effect (e.g., 0.1 nM to 100 µM).
- Apply Treatment: Apply the drug combinations to the cells. Include controls for no treatment (vehicle) and single-agent treatments for both drugs.
- Incubate: Incubate the plate for a predetermined period (e.g., 72 hours).
- Measure Response: Add the viability assay reagent according to the manufacturer's instructions and measure the signal (e.g., absorbance, luminescence).
- Data Analysis:
- Normalize the data to the vehicle control (100% viability) and a positive control for death (0% viability).
- For each drug combination, fit the data to the Hill equation: ( E = E{max} \times [D]^n / (EC{50}^n + [D]^n) ) where (E) is the effect, ( [D] ) is the drug concentration, (E{max}) is the maximal effect, (EC{50}) is the half-maximal effective concentration, and (n) is the Hill coefficient [13].
- Use software (e.g., Prism, BMDS) to perform nonlinear regression and extract the ECâ‚…â‚€ and Eₘâ‚â‚“ for the combination and individual drugs.
Protocol 2: Longitudinal Tracking of Tumor Heterogeneity via Liquid Biopsy
Objective: To monitor the clonal evolution of a tumor in response to combination therapy using circulating tumor DNA (ctDNA) from blood samples.
Materials:
- Blood collection tubes (e.g., Streck cfDNA tubes)
- cfDNA extraction kit
- Next-generation sequencing (NGS) platform (e.g., for targeted panel sequencing)
- Bioinformatics pipeline for variant calling and clonal analysis
Methodology:
- Baseline Sample Collection: Draw a blood sample from the patient prior to initiating therapy.
- On-Treatment Sampling: Collect serial blood samples at predefined time points during treatment (e.g., every cycle, at suspected progression).
- cfDNA Isolation: Process the blood samples to isolate plasma, then extract cell-free DNA (cfDNA) from the plasma.
- Library Preparation and Sequencing: Prepare NGS libraries using a panel that targets genes relevant to the cancer type and known resistance mechanisms. Sequence the libraries to high coverage.
- Bioinformatic Analysis:
- Map sequencing reads to the reference genome and call somatic variants (single nucleotide variants, indels, copy number alterations).
- Estimate the variant allele frequency (VAF) for each mutation.
- Track the changes in VAF for each mutation over time. The emergence or expansion of a clone with specific mutations indicates selection and the development of resistance [16].
- Correlation with Clinical Response: Integrate the ctDNA clonal dynamics data with the patient's radiological and clinical response to understand the drivers of treatment success or failure.
Conceptual Diagrams
DOT Code for Eco-Evolutionary Feedback in Therapy
DOT Code for Dose-Response Model Comparison
DOT Code for Tumor Heterogeneity and Therapy Resistance
The Scientist's Toolkit: Research Reagent Solutions
| Research Reagent / Tool | Function in Experimentation |
|---|---|
| Benchmark Dose Software (BMDS) | Provides a suite of statistical models for dose-response analysis and benchmark dose estimation, widely used in regulatory toxicology and risk assessment [13] [15]. |
| Method of Regularized Stokeslets (MRS) | A computational fluid dynamics method used to model locomotion and fluid-structure interactions at small scales (e.g., bacterial movement), which can inform on drug delivery dynamics [20]. |
| Immersed Boundary (IB) Method | A numerical framework for simulating fluid-structure interaction, useful for modeling biological processes like cilia-driven flow or blood flow, with applications in therapeutic distribution [20]. |
| Circulating Tumor DNA (ctDNA) Assays | Enable non-invasive, longitudinal monitoring of tumor burden and clonal evolution through blood draws, critical for assessing temporal heterogeneity and therapy response [16]. |
| Single-Cell RNA Sequencing Kits | Allow for the profiling of gene expression in individual cells within a tumor, revealing hidden heterogeneity, cell states, and potential resistance pathways not visible in bulk analyses [16]. |
| Physiologically Based Pharmacokinetic (PBPK) Modeling Software | Used to simulate the absorption, distribution, metabolism, and excretion (ADME) of drugs, helping to translate external doses into internal target tissue concentrations for more accurate dose-response modeling [15]. |
| Tecarfarin | Tecarfarin|Novel VKA Anticoagulant|For Research |
| Tenellin | Tenellin |
Technical Support Center: Troubleshooting Guides and FAQs
This section addresses common technical and strategic challenges researchers face when implementing Project Optimus principles in the development of combination therapies, with a focus on mathematical modeling approaches.
Frequently Asked Questions
FAQ 1: Our first-in-human trial did not reach a Maximum Tolerated Dose (MTD). How can we justify a dose for further development without this traditional benchmark?
- Answer: The absence of an MTD is common with modern targeted therapies. A robust justification should be based on the Totality of Evidence [21] [22]. This includes:
- Pharmacokinetic/Pharmacodynamic (PK/PD) Data: Demonstrate target saturation or engagement using validated biomarkers. Model the exposure-response relationship to identify the dose that achieves maximal biological effect [23] [1].
- Preliminary Efficacy Signals: Use early efficacy endpoints (e.g., overall response rate, effect on circulating tumor DNA) to inform dose selection [1].
- Model-Informed Drug Development (MIDD): Leverage quantitative approaches like quantitative systems pharmacology (QSP) or exposure-response modeling to integrate all available nonclinical and clinical data, predicting effective doses for later-stage trials [21] [22].
- Answer: The absence of an MTD is common with modern targeted therapies. A robust justification should be based on the Totality of Evidence [21] [22]. This includes:
FAQ 2: How do we design an efficient trial to compare multiple doses without making the study too large or costly?
- Answer: Implement adaptive and seamless trial designs [22].
- Adaptive Designs: Utilize designs that allow for pre-planned, interim analyses to drop underperforming dose arms based on efficacy or safety, thereby concentrating resources on the most promising doses [21].
- Seamless Designs: Combine traditionally distinct phases (e.g., Phase 1b and Phase 2) into a single trial, allowing for more efficient enrollment and the collection of long-term data on the selected dose(s) [22].
- Backfill Cohorts: In early-phase trials, add patients to lower dose levels ("backfilling") once safety has been established at higher doses. This efficiently generates rich PK/PD and preliminary activity data across a wider dose range [1].
- Answer: Implement adaptive and seamless trial designs [22].
FAQ 3: For a combination therapy, how can we optimize the dose of both drugs without running an unfeasible number of arms?
- Answer: This is a multidimensional problem best addressed with a model-informed, fit-for-purpose strategy [21].
- Leverage Monotherapy Data: Use well-characterized exposure-response and exposure-safety models for each individual agent to inform the combination design [22].
- Factorial Designs: Employ designs that test a limited number of dose levels for each drug in combination, using modeling to interpolate and predict outcomes for untested dose pairs [21].
- Clinical Utility Index (CUI): Use a CUI to quantitatively integrate efficacy and safety data from the combination arms, creating a composite score to objectively identify the optimal dose pair that balances benefit and risk [1].
- Answer: This is a multidimensional problem best addressed with a model-informed, fit-for-purpose strategy [21].
FAQ 4: How should we handle patient-reported outcomes (PROs) and quality-of-life data in our dose-optimization models?
- Answer: PROs are a critical component of the tolerability assessment under Project Optimus [24].
- Systematic Collection: Integrate validated PRO instruments into your clinical trials to capture symptomatic adverse events and the impact of treatment on physical function and quality of life [22].
- Quantitative Integration: Model the relationship between drug exposure and the time to deterioration or improvement in PRO scores. This data can be incorporated into a Clinical Utility Index alongside traditional efficacy and safety endpoints to provide a patient-centric view of the optimal dose [24] [22].
- Answer: PROs are a critical component of the tolerability assessment under Project Optimus [24].
Quantitative Data on Dose Optimization
The following tables summarize key quantitative findings and methodological approaches relevant to dose optimization.
Table 1: Evidence for the Need of Improved Dose Optimization in Oncology
| Data Point | Finding | Source / Context |
|---|---|---|
| Dose Modification Rate | 48% of patients in Phase 3 trials of molecularly targeted agents required dose modifications from the recommended dose. | Analysis of tolerability in phase 3 trials [23] |
| Post-Marketing Dose Reevaluation | The FDA has required additional studies to re-evaluate the dosing of over 50% of recently approved cancer drugs. | FDA observations on recent approvals [1] |
| Dose Reduction/Interruption | Registration trials for new oral targeted agents (2010-2020) showed median dose reduction and interruption rates of 28% and 55%, respectively. | Review of 59 newly approved oral molecular entities [21] |
Table 2: Key Model-Informed Drug Development (MIDD) Approaches for Dose Optimization
| Modeling Approach | Primary Function | Application in Combination Therapy |
|---|---|---|
| Exposure-Response (E-R) Modeling | Correlates drug exposure (e.g., AUC, C~trough~) with efficacy or safety endpoints to predict the response at different doses. | Can be developed for each drug in a combination to understand their individual and potentially synergistic contributions. |
| Quantitative Systems Pharmacology (QSP) | Incorporates biological mechanisms to predict drug effects. Uses limited clinical data to understand complex interactions. | Highly valuable for simulating the interaction between two drugs and identifying dose regimens that maximize synergy and minimize overlapping toxicities [22]. |
| Clinical Utility Index (CUI) | A quantitative framework that creates a composite score by integrating multiple endpoints (efficacy, safety, PROs) to rank different doses. | Ideal for objectively selecting the optimal dose pair from a combination trial by balancing the efficacy and safety profiles of both agents [1]. |
| Population PK (PopPK) Modeling | Describes the pharmacokinetics and sources of variability in a patient population. | Can identify covariates (e.g., organ function, drug-drug interactions) that may necessitate dose adjustments in a combination setting [22]. |
Experimental Protocols for Dose Optimization
Protocol 1: Randomized Dose Comparison in an Early-Phase Trial
Objective: To select the optimal dose for registrational trials by comparing at least two doses for efficacy and safety.
Methodology:
- Dose Selection: Based on Phase 1a data, select two or more doses for head-to-head comparison. The doses should be sufficiently spaced (e.g., 2-3 fold apart in exposure) and could include the estimated minimum biologically effective dose (MBED) and the highest tolerable dose [23] [1].
- Study Population: Enroll patients from the intended later-phase population. Expansion cohorts in Phase 1 can be used for this randomization.
- Randomization: Randomize patients to the selected dose arms. The study does not need to be powered for a formal statistical superiority comparison but must be sufficiently sized to characterize the shape of the dose-response and dose-toxicity relationships [23].
- Endpoint Assessment: Collect comprehensive data on:
- Efficacy: Primary efficacy endpoints (e.g., ORR) and relevant biomarker data (e.g., ctDNA) [1].
- Safety: Incidence of adverse events, dose reductions, interruptions, and discontinuations.
- PK/PD: Intensive sampling for exposure assessment and target engagement biomarkers.
- PROs: Patient-reported outcomes on symptoms and quality of life [22].
- Data Integration & Dose Selection: Analyze the totality of evidence. Use a Clinical Utility Index (CUI) or similar quantitative framework to integrate the efficacy and safety data and select the dose with the most favorable benefit-risk profile for the registrational trial [1].
Protocol 2: Model-Informed Dose Optimization for a Combination Therapy
Objective: To identify the optimal dose pair for two investigational drugs (Drug A and Drug B) used in combination using quantitative modeling.
Methodology:
- Prior Knowledge: Develop robust PopPK and exposure-response models for Drug A and Drug B as monotherapies using all available nonclinical and clinical data [22].
- Combination Trial Design: Initiate a clinical trial testing a limited set of dose pairs (e.g., two dose levels of Drug A combined with two dose levels of Drug B).
- Data Collection: Collect rich PK, PD, efficacy, and safety data from all arms of the combination trial.
- Model Development & Simulation:
- Develop a QSP model that incorporates the mechanisms of action of both drugs and their potential interactions [22].
- Calibrate and verify the model using the clinical data from the combination trial.
- Use the verified model to simulate a wide range of untested dose pairs for Drug A and Drug B, predicting key efficacy and safety outcomes for each virtual pair.
- Dose Selection: Identify the simulated dose pair that maximizes predicted efficacy while maintaining a tolerable safety profile. This model-informed recommendation can then be validated in a subsequent expansion cohort or registrational trial.
Visualizing the Project Optimus Workflow and Modeling Strategy
Project Optimus vs Legacy Dose Finding
MIDD Toolkit for Dose Optimization
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials and Tools for Project Optimus-Aligned Research
| Tool / Reagent | Function in Dose Optimization | Application Example |
|---|---|---|
| Validated PD Biomarker Assays | To quantitatively measure target engagement and biological effect of the drug at different dose levels. | Immunoassays or flow cytometry to confirm receptor occupancy or modulation of a downstream signaling pathway, helping to define the minimum biologically effective dose (MBED) [23] [1]. |
| LC-MS/MS Systems | For high-sensitivity quantification of drug and metabolite concentrations in biological matrices (plasma, tissue) to support robust PK analysis. | Generating concentration-time data for population PK modeling, which is foundational for all exposure-response analyses [22]. |
| Circulating Tumor DNA (ctDNA) Assays | To serve as an early, dynamic biomarker of tumor response and resistance. | Tracking changes in ctDNA levels in response to different doses in early-phase trials to inform efficacy signals before traditional radiological assessments [1]. |
| Modeling & Simulation Software | Platforms for performing complex quantitative analyses, including population PK, exposure-response, and QSP modeling. | Using software like R, NONMEM, or specialized QSP platforms to integrate all data sources, simulate untested doses, and identify the optimal dose with a superior benefit-risk profile [21] [22]. |
| Validated Patient-Reported Outcome (PRO) Instruments | To systematically capture the patient's perspective on treatment tolerability and impact on quality of life. | Integrating PRO data into safety assessments and the Clinical Utility Index to ensure the selected dose is not only effective but also tolerable from the patient's viewpoint [24] [22]. |
| APN-C3-NH-Boc | APN-C3-NH-Boc|Alkyl/Ether PROTAC Linker | APN-C3-NH-Boc is an alkyl/ether PROTAC linker with an alkyne handle for click chemistry. For Research Use Only. Not for human use. |
| THK-523 | THK-523, CAS:1573029-17-0, MF:C17H15FN2O, MW:282.32 | Chemical Reagent |
The Modeler's Toolkit: Key Approaches and Clinical Applications in Combination Therapy
This technical support center provides troubleshooting guides and FAQs for researchers using mathematical modeling to optimize combination therapy doses. The guidance is framed within the context of a broader thesis on this topic.
Frameworks for Combination Therapy Optimization
The table below summarizes the core modeling frameworks used in therapeutic research.
| Modeling Framework | Core Principle | Advantages for Combination Therapy | Key Challenges | Representative Applications |
|---|---|---|---|---|
| Ordinary Differential Equations (ODE) | Represents system states as continuous variables changing over time via differential equations. [25] | Well-established for PK/PD; efficiently describes drug concentration and effect. [26] | Difficult to capture spatial heterogeneity and individual entity history. [26] | Modeling pharmacokinetics and signaling pathways (e.g., NF-κB, STAT3). [25] [27] |
| Agent-Based Modeling (ABM) | Models system from the bottom-up through interactions of discrete, autonomous agents. [26] | Naturally captures tumor heterogeneity, spatial effects, and cell-cell interactions. [26] | High computational cost; parameterization and validation can be complex. [26] | Simulating tumor-immune cell interactions and therapy responses in the tumor microenvironment. [26] |
| Multiscale Modeling | Integrates multiple models operating at different biological scales (e.g., molecular, cellular, tissue). [28] | Mechanistically links drug pharmacokinetics to cellular and tissue-level responses. [28] | Designing scale-coupling functions; high computational complexity. [29] | Predicting in vivo efficacy of CAR-T cell therapies in solid tumors. [28] |
| TJ191 | TJ191|Selective Anti-Cancer Small Molecule|RUO | TJ191 is a potent cytostatic/cytotoxic agent for T-cell leukemia/lymphoma research. It targets cells with low TβRIII. For Research Use Only. Not for human use. | Bench Chemicals | |
| Tos-PEG4-acid | Tos-PEG4-acid, MF:C16H24O8S, MW:376.4 g/mol | Chemical Reagent | Bench Chemicals |
Frequently Asked Questions
ODE Model Troubleshooting
Q: My ODE model of a pro-/anti-inflammatory signaling pathway fails to resolve inflammation. What could be wrong?
A: A lack of resolution often points to missing negative feedback loops. Ensure your model includes key regulatory components. For instance, in a macrophage polarization model, the inclusion of the SOCS (Suppressor of Cytokine Signaling) family of proteins is critical. SOCS1 and SOCS3 act as part of negative feedback loops that resolve both the M1 and M2 pathways. SOCS3 inhibits the transcription of TNFα mRNA, and both SOCS1 and SOCS3 inhibit the activation of STAT3. Without these, the model may not definitively resolve the inflammatory response. [25]
Q: How can I improve the generalizability of my PK-ODE model to predict untested dosing regimens?
A: Consider moving beyond traditional nonlinear mixed-effects models. A cutting-edge approach is to use Neural Ordinary Differential Equations (Neural-ODE). This method uses a neural network to learn the dynamics of the ODE system directly from data. Studies have shown that Neural-ODE models demonstrate superior performance in predicting pharmacokinetic profiles for dosing regimens that were not part of the training data, a common limitation of other machine learning and traditional PK models. [30]
Agent-Based Model Troubleshooting
Q: I am getting a "Type mismatch: cannot convert from [TYPEA] to [TYPEB]" error in my agent-based model. How do I fix it?
A: This is a common compile-time error where a variable is assigned a value of an incorrect type. [31] For example, a variable defined as a double (a decimal number) might be used in a context that requires a boolean (true/false) value, such as the condition for a SelectOutput block.
- Solution: Carefully check the defined type of the variable causing the error and the type expected by the function or block where it is used. Correct the type declaration or the value assignment to ensure they match. [31]
A: This is a runtime error related to the model's logic in a discrete-event flowchart. It indicates that an agent (e.g., a cell) has reached a port (an exit point) in a block, but there is no connected block for the agent to move to next. [31]
- Solution: Review your flowchart connectivity. Ensure the port in question is connected to a subsequent block, such as a
DelayorSinkblock, to handle the agent. [31]
Multiscale Model Troubleshooting
Q: What is the most efficient design pattern for coupling different simulators in a multiscale model?
A: A robust software science co-design pattern uses five modules: one launcher, two simulators, and two transfer modules. Each transfer module contains an interface for receiving data, an interface for sending data, and a critical transformation process. The transformation process is responsible for converting the data from one scale into a format that is meaningful at the other scale (e.g., converting population-level average signals to individual cell stimuli and vice versa). This design separates scientific (the transformation logic) from technical (data exchange) concerns, improving efficiency and maintainability. [29]
Q: Our multiscale model of CAR-T cell therapy in solid tumors is not showing efficacy in virtual patients. What factors should we investigate?
A: The lack of efficacy in silico can reveal critical biological barriers. Use your model for sensitivity analysis to identify the most influential parameters. Key factors to investigate include:
- CAR-T cell tumor infiltration: The insufficient presence of CAR-T cells in the tumor tissue is a major barrier. [28]
- Tumor antigen heterogeneity: Variable expression of the target antigen can lead to immune escape. [28]
- Immunosuppressive Tumor Microenvironment (TME): Factors like regulatory T cells, myeloid-derived suppressor cells, and immunosuppressive cytokines can inactivate CAR-T cells. [28]
- CAR-T product parameters: Explore the impact of CAR affinity, CAR density on the T cell surface, and the ratio of CD4+ to CD8+ T cells in the infused product. [28]
Experimental Protocols & Workflows
Protocol 1: Building a QSP Model for CAR-T Cell Therapy
This protocol outlines the steps for developing a Multiscale Quantitative Systems Pharmacology (QSP) model to predict the efficacy of CAR-T therapies in solid tumors. [28]
- Define Model Scope and Biology: Identify key actors: CAR-T cells (CD4+, CD8+), tumor cells with antigen expression levels, and major components of the tumor microenvironment (e.g., immunosuppressive cells).
- Formulate the Mathematical Framework:
- Use ODEs to describe the cellular kinetics of CAR-T cells (expansion, contraction, persistence).
- Use ABM rules or PDEs to capture spatial distribution and cell-cell contact dynamics within the tumor.
- Integrate a PK/PD model for any co-administered drugs in a combination therapy.
- Calibrate with Multimodal Data:
- In vitro: Use data from co-culture assays of CAR-T cells and tumor cells to calibrate killing kinetics and cytokine release.
- In vivo: Use animal model data (e.g., murine xenografts) to calibrate CAR-T biodistribution, tumor volume dynamics, and overall survival.
- Generate a Virtual Patient Population: Create a virtual cohort by sampling key system parameters (e.g., tumor burden, antigen expression level, immune contexture) from distributions informed by clinical data.
- Simulate and Optimize Dosing: Run prospective simulations with the virtual patients to test different dosing strategies, such as flat dosing versus step-fractionated dosing, to identify regimens that maximize efficacy and minimize toxicity. [28]
Protocol 2: Co-Simulation of Brain Models Across Scales
This protocol describes a workflow for co-simulating a macroscopic brain network model with a microscopic spiking neural network. [29]
- Setup Simulators: Establish two specialized simulators: The Virtual Brain (TVB) for the whole-brain network model and NEST for the spiking neural network model of a specific region (e.g., hippocampus CA1).
- Implement the Co-Design Pattern:
- The Launcher starts both simulators and handles global coordination.
- Simulator A (TVB) runs its simulation for a short time window.
- Transfer Module A→B receives the macroscopic output from TVB (e.g., average neural population activity). Its transformation process converts this into input for the micro-scale model (e.g., synaptic currents to individual neurons).
- Simulator B (NEST) runs its simulation for the same time window, using the transformed input.
- Transfer Module B→A receives the microscopic output from NEST (e.g., aggregated spiking activity). Its transformation process converts this back into a format for the macro-scale model (e.g., an input current to the neural mass model in TVB).
- Validate with Multiscale Data: Compare the co-simulation output with empirical data that spans scales, such as simultaneous electrocorticography (ECoG) and local field potential (LFP) recordings. [29]
Research Reagent Solutions
The table below lists key computational tools and platforms used in advanced pharmacological modeling.
| Tool / Platform | Type | Primary Function in Research |
|---|---|---|
| Stan (with CmdStanR) [27] | Statistical Inference Engine | Bayesian parameter estimation for complex ODE models, such as pharmacokinetic models. |
| AnyLogic [31] | Commercial Modeling Platform | Integrated environment for developing agent-based, discrete-event, and system dynamics models. |
| The Virtual Brain (TVB) [29] | Open-Source Platform | Simulation of whole-brain network dynamics based on individual neuroimaging-derived connectomes. |
| NEST [29] | Open-Source Simulator | Simulation of large-scale spiking neural network models at the level of individual neurons and synapses. |
| Neural-ODE [30] | Machine Learning Method | Learning the structure and parameters of differential equation systems directly from time-series data. |
Model Diagrams and Workflows
ODE Model of Macrophage Polarization
Multiscale Co-Simulation Design Pattern
ABM Model for Tumor-Immune System
Leveraging Exposure-Response and Quantitative Systems Pharmacology (QSP) Models
Frequently Asked Questions (FAQs)
Q1: Do QSP models require vast amounts of data to be built? No. While building a QSP model from scratch requires data to inform its parameters, using pre-existing, literature-based models for well-understood systems (e.g., renal function, bone metabolism) can significantly reduce data requirements. You primarily need pharmacokinetic (PK) data and information on the drug's mechanism of action or biomarkers. [32]
Q2: Is QSP modeling accepted by regulatory agencies like the FDA? Yes. QSP is increasingly used to support Investigational New Drug (IND), New Drug Application (NDA), and Biologics License Application (BLA) submissions. Submissions incorporating QSP models have been rising, and they have been used, for instance, to evaluate dosing regimens in regulatory submissions. [32]
Q3: What is the main difference between QSP and traditional population PK/PD (popPK/PD) models? PopPK/PD models typically describe the empirical relationship between plasma drug concentrations and a pharmacodynamic effect. In contrast, QSP models mechanistically describe the relationship between drug concentrations at the site of action and the resulting effects, accounting for complex biological networks, multiple sequential processes, endogenous substrates, and feedback mechanisms. This makes QSP particularly valuable for simulating combination therapies with different mechanisms of action. [32]
Q4: How can I have confidence in a QSP model's predictions, especially with many uncertain parameters? Using Virtual Populations (VPs) is a key method for assessing confidence. By running simulations across a family of parameter sets, you can generate a distribution of predictions. You can then quantify the robustness of a qualitative prediction (e.g., a drug-scheduling effect) by determining in what proportion of virtual population simulations the effect persists, compared to a null hypothesis. [33]
Q5: Can QSP models be built if the drug's mechanism of action is not fully understood? Yes. Gaps in knowledge can be addressed by hypothesizing a mechanism, checking the model against available data, and iteratively refining it with new experimental results. Literature searches for similar compounds and collaboration with expert consultants are crucial for filling these gaps. [32]
Q6: At what stage of drug development can QSP be applied? QSP can add value at all stages, from early discovery to late-stage development. Early on, it can aid target validation and candidate selection. Later, it can optimize clinical trial design, evaluate subpopulations, and support dosage decisions for registrational trials without the need for new clinical studies. [32] [34]
Troubleshooting Common QSP Challenges
The table below outlines common technical challenges encountered during QSP modeling and practical solutions to address them.
| Challenge | Description & Potential Solutions |
|---|---|
| Parameter Identifiability & Estimation | Description: It can be difficult to uniquely estimate a large number of parameters in complex models, leading to uncertainty. [35]Solutions:• Use profile likelihood methods to check if parameters are practically identifiable. [35]• Employ Markov Chain Monte Carlo (MCMC) approaches to explore the posterior distribution of parameters and identify those with wide, unconstrained distributions. [35] |
| Model Validation for Qualitative Predictions | Description: Standard pharmacometric validation methods (e.g., goodness-of-fit plots) are not always suitable for assessing QSP models designed for qualitative, systems-level predictions. [33]Solutions:• Use Virtual Populations to generate distributions of predictions and statistically quantify the robustness of qualitative findings (e.g., "in 95% of simulations, the sequential regimen was superior"). [33] |
| Balancing Granularity & Complexity | Description: Determining the right level of biological detail ("granularity") is difficult. Too much detail makes the model complex and slow; too little reduces predictive power. [35]Solutions:• Let the research question guide the required granularity. [35]• Use model reduction techniques to lump variables and simplify large network models where possible. [35] |
| Integrating Disparate Data Sources | Description: QSP models are often constrained by data from multiple sources (in vitro, in vivo, clinical) and scales (molecular, cellular, organ), which can be heterogeneous. [33]Solutions:• The model itself serves as a framework to integrate this multi-scale data. [34] A collaborative, iterative cycle with experimental labs is essential to fill knowledge gaps and refine the model. [35] |
Featured Experiment: Optimizing a Breast Cancer Combination Immunotherapy Regimen
This section details a specific research experiment that leveraged mathematical modeling to optimize doses for combination therapy, directly supporting the thesis context.
Experimental Objective
To develop and validate a mathematical model that optimizes the medication regimen for combining an mRNA-based cancer vaccine with anti-CTLA-4 antibody therapy for breast cancer, aiming to maximize tumor growth inhibition while minimizing immunotoxic side effects. [36]
Detailed Methodology
Model Development: A mathematical model was constructed to describe the interactions between the mRNA-based vaccine, anti-CTLA-4 antibodies, and the tumor immune microenvironment. The model likely includes components for immune cell activation, tumor cell killing, and inhibitory signaling via CTLA-4. [36]
Parameter Estimation: The model was parameterized using experimental data. The Markov Chain Monte Carlo (MCMC) method was employed to estimate model parameters, a robust approach for dealing with parameter uncertainty in complex biological models. [36]
Model Simulation & Validation: Simulations from the parameterized model were compared against experimental results not used in the training phase to assess the model's predictive capability and build credibility. [36]
Regimen Optimization: The gradient descent method, an optimization algorithm, was designed and applied to the validated model. This algorithm systematically adjusted the dosing variables (timing and amount) to find the regimen that best achieved the dual goals of inhibiting tumors and reducing side effects. [36]
- The optimized regimen dictated that the anti-CTLA-4 antibody should be administered after the vaccination. [36]
- Within a safe range, the dose of the antibody should positively correlate with the dose of the vaccine. [36]
- The study provides a theoretical basis for selecting combination therapy regimens in clinical trials, moving beyond trial-and-error approaches. [36]
Logical Workflow Diagram
The following diagram illustrates the sequential, iterative workflow of the featured QSP experiment for optimizing combination therapy.
Research Reagent Solutions & Essential Materials
The table below lists key computational and methodological "reagents" essential for conducting QSP research like the featured experiment.
| Item | Function in Research |
|---|---|
| Markov Chain Monte Carlo (MCMC) | A computational algorithm for estimating parameters in complex models, especially when facing uncertainty. It explores the probability distribution of parameters given the data. [36] |
| Gradient Descent Method | An optimization algorithm used to find the minimum of a function. In this context, it was designed to find the dosing regimen that minimizes tumor size and side effects. [36] |
| Virtual Populations (VPs) | A family of model parameter sets used to account for uncertainty and biological variability. VPs generate distributions of predictions, allowing researchers to quantify the robustness of results. [33] |
| Pre-Validated QSP Model Libraries | Existing models for specific biological systems (e.g., cardiac action potential, liver disease) that can be adapted for new projects, saving significant time and resources compared to building from scratch. [32] [37] |
| Model Credibility Assessment Framework | A set of criteria (e.g., from the ASME or EMA) used to evaluate the credibility of computational models for a specific context of use, which is critical for regulatory submissions. [38] |
FAQs: Foundational Concepts in In Silico Therapy Design
Q1: What is the core value of using mathematical models in CAR-T and Targeted Radionuclide Therapy (TRT) development?
Mathematical models provide a systematic and quantitative framework to understand the complex, dynamic interactions between therapy and cancer, which are often difficult or costly to probe experimentally [39] [40]. They enable researchers to simulate treatment outcomes in silico, offering a resource-saving method to test hypotheses, optimize dosing schedules, and personalize treatment protocols before moving to clinical trials [39] [5] [41]. For combination therapies, models are crucial for determining the optimal timing and sequence of treatments [42].
Q2: What are the primary types of computational models used in this field, and when should I use them?
The choice of model depends on the research question and the scale of the biological process being investigated.
| Model Type | Key Characteristics | Best Use Cases |
|---|---|---|
| Agent-Based Models (ABM) | Simulates actions and interactions of autonomous entities (e.g., individual cells) in a spatial environment to explore emergent system behavior [39]. | Studying the effects of spatial heterogeneity, cell-cell contact interactions, and the emergence of resistant cell populations [39]. |
| Ordinary Differential Equation (ODE) Models | Describes system dynamics through equations that define the rates of change of population-level quantities (e.g., tumor cell count, CAR-T cell count) over time [41] [42]. | Modeling bulk population dynamics, pharmacokinetics/pharmacodynamics (PK/PD), and predicting overall tumor burden [41] [42]. |
| Pharmacokinetic-Pharmacodynamic (PKPD) Models | A class of ODE models that specifically links the pharmacokinetics (what the body does to the drug) to the pharmacodynamic response (what the drug does to the body) [41]. | Predicting the relationship between drug/CAR-T affinity, antigen abundance, tumor cell depletion, and therapy expansion [41]. |
| Monte Carlo Simulations | Uses random sampling to model the probability of different outcomes in processes that are inherently stochastic [43]. | Simulating radiation track structures and calculating the precise number and complexity of DNA damage events caused by radionuclides at a cellular level [43]. |
Q3: What key biological determinants does modeling suggest are critical for CAR-T cell therapy success?
Computational studies have highlighted several critical factors:
- CAR-T Cell Persistence and Expansion: The ability of CAR-T cells to survive and proliferate in vivo is a major determinant of long-term remission and preventing relapse [41].
- Tumor Antigen Heterogeneity: Intratumoral heterogeneity in antigen expression is a leading cause of therapeutic resistance. Models show that cells with low antigen expression can form a "shield," protecting high-antigen cells from eradication [39].
- Tumor Proliferation Rate: The intrinsic growth rate of a tumor is a key parameter in scheduling combination therapies, with faster-proliferating tumors requiring different timing between TRT and CAR-T administration [42].
Q4: How can in silico models help overcome antigen escape in CAR-T therapy for solid tumors?
Models are used to design and test strategies to overcome antigen escape, a phenomenon where tumor cells stop expressing the target antigen. A prominent strategy is multi-antigen recognition, such as syn-Notch receptors, where an engineered receptor induces expression of a CAR upon recognition of a primary antigen, creating T-cells that can target two different antigens [39]. While powerful, models also highlight that this approach can increase the risk of on-target, off-tumor toxicity, necessitating careful dosimetry [39].
Q5: What are the principal considerations for optimizing Targeted Radionuclide Therapy (TRT) based on modeling?
Mathematical models of TRT emphasize several optimization principles [44]:
- Nuclide-to-Antibody Ratio: The density of radioconjugates on cancer cells determines radiation energy deposition. A low ratio or an excess of unlabeled antibodies can saturate receptors and mitigate cancer cell damage.
- Cancer Binding Capacity-Based Dosing: Doses significantly exceeding the total number of target receptors on cancer cells should be avoided, as the excess circulates in the bloodstream, contributing to toxicity without enhancing efficacy.
- Particle Range-Guided Multi-Dosing: For short-range alpha emitters, an initial dose to saturate cancer cells allows subsequent doses to target viable cells that continue expressing receptors, improving dose redistribution.
Troubleshooting Guides
Guide 1: Addressing Inadequate Tumor Control in CAR-T Cell Therapy Simulations
Problem: Your model predicts poor tumor control or early relapse after CAR-T cell therapy.
| Possible Cause | Diagnostic Checks | Potential Solutions |
|---|---|---|
| Low CAR-T Cell Persistence | Check the simulated dynamics of activated vs. non-activated CAR-T cells over time. Is the population declining rapidly? [41] | Model the administration of "next-generation" CARs (e.g., 4th gen TRUCKs) that include cytokine genes to enhance persistence and memory formation [40]. |
| High Antigen Heterogeneity | Analyze the spatial distribution and phenotypic evolution of tumor cell clones, particularly those with low antigen expression [39]. | Simulate a switch to a multi-antigen targeting strategy (e.g., syn-Notch receptor circuits) to overcome heterogeneity and antigen escape [39]. |
| Suboptimal Dosing | Run sensitivity analyses on the initial CAR-T cell dose and the killing rate parameter (k1 in ODE models) [39] [41]. |
Test multiple dosing regimens or model combination therapy with TRT to target antigen-negative cells via bystander effects [39] [42]. |
| CAR-T Cell Exhaustion | Incorporate an "exhausted" state in your model and track its population. Check if the rate of exhaustion upon tumor cell encounter (k2) is too high [42]. |
Investigate the effect of costimulatory domains in your CAR design (e.g., 4-1BB vs. CD28) within the model, as these can influence exhaustion profiles [40]. |
Guide 2: Managing Toxicity and Off-Target Effects in TRT
Problem: Your TRT model shows effective tumor kill but unacceptably high toxicity to healthy tissues, particularly bone marrow.
| Possible Cause | Diagnostic Checks | Potential Solutions |
|---|---|---|
| Excessive Unanchored Radionuclides | Quantify the ratio of radionuclides bound to cancer cells versus those circulating freely in the bloodstream over time [44]. | Optimize the injected dose to match the tumor's binding capacity, avoiding large excesses that remain in circulation [44]. |
| Suboptimal Radionuclide Choice | Compare the simulated DNA damage (e.g., DSB/Gbp/decay) and effective range of different radionuclides in your model [43]. | For small tumors/micrometastases, model switching from a beta emitter (e.g., ¹â·â·Lu) to a short-range alpha emitter (e.g., ²²âµAc), which deposits more energy over a smaller distance, sparing surrounding tissues [43]. |
| Inadequate Dosing Schedule | Simulate the cumulative dose to dose-limiting organs (e.g., bone marrow) for single vs. fractionated dosing schedules. | Implement dose fractionation. Splitting the total dose into several smaller administrations can reduce peak toxicity and allow healthy tissue recovery [44]. |
Experimental Protocols & Workflows
Protocol 1: Building an Agent-Based Model (ABM) to Study CAR-T Therapy Against Heterogeneous Tumors
This protocol is based on the in silico study of tumor-derived organoids [39].
1. Define the Simulation Environment and Initial Conditions:
- Set up a 3D grid (e.g., 1000x1000x1000 µm) with a spherical organoid seeded at the center.
- To model heterogeneity, assign each tumor cell a mutant oncoprotein value drawn from a normal distribution (e.g., mean=1, SD=0.25, range 0-2). Discretize cells into types based on this value (e.g., Type 1: 1.5-2.0, Type 2: 1.0-1.5, etc.) [39].
2. Program Agent Behaviors and Rules:
- Tumor Cells: Proliferation rate and immunogenicity should scale proportionally with the oncoprotein value
o. Set a threshold (e.g., o < 0.5) below which cells are not recognized by CAR-T cells [39]. - CAR-T Cells: Program rules for random motility, activation upon contact with a tumor cell (if
o> threshold), cytotoxic killing, and proliferation post-activation.
3. Implement Therapy and Run Simulations:
- Introduce a defined dose of CAR-T cells at a specific time point (e.g., day 7).
- Run simulations for different T-cell-to-cancer ratios and dosing strategies (single vs. multiple doses).
4. Analyze Outputs:
- Track total tumor cell count and mean oncoprotein expression over time.
- Observe spatial structures, such as the formation of a "shield" of low-antigen cells protecting high-antigen cells [39].
- Quantify the number of "free" CAR-T cells (those that have not engaged a target) as a proxy for potential side-effect risk.
ABM Workflow for CAR-T Therapy
Protocol 2: Implementing a Combined ODE Model for TRT and CAR-T Combination Therapy
This protocol is based on the work combining two previously published models [42].
1. Define Model Variables and Equations: The model typically tracks these populations:
N_T: Non-irradiated tumor cellsN_R: Irradiated tumor cellsN_C: CAR-T cells
The system of ODEs can be structured as follows [42]:
- dNT/dt = ÏNT - H(t-Ï„TRT) * kRxT * NT - H(t-Ï„CAR) * k1 * NT * NC (Change in non-irradiated cells = Growth - TRT effect - CAR-T killing)
- dNR/dt = H(t-Ï„TRT) * kRxT * NT - H(t-Ï„CAR) * k1 * NR * NC - kcl * N_R (Change in irradiated cells = TRT effect - CAR-T killing - Clearance)
- dNC/dt = k2 * (NT + NR) * NC - H(t-τTRT) * kRxC * NC - θ * NC (Change in CAR-T cells = Proliferation - TRT effect - Death)
2. Parameterize the Model:
- TRT Parameters (
k_Rx_T,k_Rx_C,k_cl): Obtain from preclinical TRT studies. For alpha emitters like ²²âµAc, the radiation effect termk_Rxcan be modeled using a linear-quadratic equation with a dose protraction factor [42]. - CAR-T Parameters (
k_1,k_2,θ): Estimate from mouse models.k_1is the killing rate,k_2is the proliferation rate upon tumor encounter, andθis the death rate [42]. - Tumor Parameter (
Ï): Fit from control group tumor growth data.
3. Simulate Combination Therapy:
- Use the Heaviside function
H(t-τ)to turn treatments on at specific timesτ_TRTandτ_CAR. - Run simulations for different sequences (TRT first vs. CAR-T first) and intervals between therapies.
4. Identify Optimal Scheduling:
- The tumor proliferation rate
Ïis a critical parameter. Models suggest that for faster-proliferating tumors, the interval between TRT and CAR-T should be shorter [42].
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Research | Application Context |
|---|---|---|
| Geant4-DNA Toolkit | A Monte Carlo simulation toolkit for modeling particle track structures, water radiolysis, and biological damage induced by ionizing radiation at the DNA level [43]. | Used to simulate and compare DNA damage (e.g., DSB yields) from different TRT radionuclides (¹â·â·Lu, ²²âµAc, ²¹²Pb) and source localizations [43]. |
| CS1-CAR-T Cells | Chimeric Antigen Receptor T cells engineered to target the CS1 antigen, which is expressed in multiple myeloma cells [42]. | Used in preclinical mouse models of multiple myeloma to parameterize ODE models for CAR-T cell killing rate (k1) and persistence (θ) [42]. |
| ²²âµAc-DOTA-daratumumab | An alpha-emitting radioconjugate. Daratumumab (anti-CD38 antibody) targets multiple myeloma cells, delivering ²²âµAc to the tumor site [42] [44]. | Critical for parametrizing and validating TRT models. Used to study the impact of labeling ratio, dosing, and schedule on efficacy and toxicity [44]. |
| Syn-Notch Receptor System | An engineered receptor system that induces expression of a CAR upon recognition of a primary tumor antigen, enabling multi-antigen targeting [39]. | Modeled in silico to design and test strategies to overcome antigen escape and heterogeneity in solid tumors [39]. |
| 5th Generation CARs | CAR designs that include a truncated cytoplasmic domain of cytokine receptors (e.g., IL-2R) to incorporate JAK-STAT signaling, enhancing persistence and resistance to immunosuppression [40]. | Modeled computationally to improve CAR-T cell performance in challenging environments like solid tumors by providing a more complete T cell activation signal [40]. |
Signaling Pathways & Therapy Logic
CAR-T Cell Signaling and Activation Pathway
CAR-T Cell Signaling Pathway
This diagram illustrates the structural and signaling evolution of Chimeric Antigen Receptors. The core signaling involves antigen binding leading to CD3ζ phosphorylation (Signal 1) and costimulatory signaling (Signal 2). In the more advanced 5th generation CARs, an additional cytokine receptor fragment is included to incorporate JAK-STAT signaling (Signal 3), promoting enhanced persistence and memory formation, which is particularly relevant for the challenging solid tumor microenvironment [40].
Technical Support Center: Troubleshooting Guides and FAQs
Frequently Asked Questions (FAQs)
Q1: What is Model-Informed Precision Dosing (MIPD) and how does it improve upon traditional Therapeutic Drug Monitoring (TDM)?
Model-Informed Precision Dosing (MIPD) is an advanced quantitative approach that integrates mathematical and statistical models of drugs and diseases with individual patient characteristics to tailor drug dosing [45] [46]. It moves beyond traditional TDM by not only using drug concentration measurements but also incorporating patient-specific factors (e.g., demographics, genetics) and different sources of variability to predict optimal dosing regimens [45]. While TDM relies on measured drug levels to reactively adjust doses, MIPD uses models to proactively predict the best dose for an individual, increasing the safety and efficacy of pharmacological treatments [46].
Q2: What are the primary applications of MIPD in oncology and combination therapies?
In oncology, MIPD and mathematical modeling are used to move beyond the traditional Maximum Tolerated Dose (MTD) paradigm [5]. They help optimize treatment strategies for new therapeutics like targeted therapies and immunotherapies, whose efficacy can saturate, making the MTD approach suboptimal [5]. Key applications include:
- Personalizing Treatment Schedules: Using models to determine optimal drug dosing, timing, and sequences for combinations [5].
- Managing Drug Resistance: Creating models that capture evolutionary dynamics to design therapies that suppress the emergence of resistance [5] [47].
- Informing Clinical Trials: Using clinical trial simulations based on drug-trial-disease models to inform trial design, duration, and response measures [48].
Q3: What software and computational tools are essential for implementing MIPD?
Implementing MIPD requires specialized software for model development, simulation, and Bayesian forecasting.
- Bayesian Forecasting Tools: Software like the open-source R package
Posologyris used for Bayesian parameter estimation and dose individualization. These tools are crucial for forecasting individualized dosing in patients enrolled in TDM programs [46]. - Professional Software: Tools like NONMEM and Monolix are industry standards for population pharmacokinetic (popPK) and pharmacodynamic (PD) model development [46].
- Regulatory-Endorsed Approaches: The FDA's Model-Informed Drug Development (MIDD) Paired Meeting Program provides a framework for discussing and validating these quantitative approaches in drug development [48].
Q4: What are common challenges when developing a pharmacokinetic/pharmacodynamic (PK/PD) model for drug combinations?
A primary challenge is capturing the complex, multi-scale dynamics of the tumor and its microenvironment, especially when combining drugs with different mechanisms of action [5]. This includes:
- Data Limitations: Models are often limited by the amount of patient-specific data available, necessitating the use of prior knowledge from preclinical and retrospective clinical data [5].
- Model Qualification: Rigorous model qualification and validation are required before clinical application. This involves assessing model risk, which considers the influence of model predictions and the potential consequence of an incorrect decision [45] [48].
- Handling Variability: Accounting for high inter- and intra-individual variability in drug PK behavior, which is common with drugs like tyrosine kinase inhibitors (TKIs) [46].
Troubleshooting Common Experimental and Modeling Issues
Issue 1: Poor Predictive Performance of a PopPK Model
- Problem: A developed population pharmacokinetic (popPK) model does not accurately predict drug concentrations in a new patient cohort.
- Solution:
- Re-evaluate Covariates: Identify and incorporate missing patient-specific covariates that influence drug clearance or volume of distribution. Common covariates include renal function (creatinine clearance), body weight, and co-medications [46].
- External Validation: Test the predictive performance of your model against an external dataset not used for model building. The FDA recommends this as part of a model risk assessment [48].
- Model Selection: Compare several published popPK models for your drug. Research has shown that for drugs like infliximab, some published models have better predictive performance than others and are more suitable for MIPD approaches [46].
Issue 2: Designing an Adaptive Therapy Schedule Based on Evolutionary Dynamics
- Problem: Difficulty in determining when to switch or pause drugs in an adaptive therapy regimen designed to control tumor evolution.
- Solution:
- Define a Dynamic Trigger: Establish a model-informed biomarker threshold for treatment decisions. For example, a schedule might involve switching from a "first-strike" to a "second-strike" therapy when the model predicts the emergence of a resistant subpopulation [5].
- Use Clinical Trial Data: Refer to existing adaptive therapy trials for guidance. For instance, trials for metastatic castration-resistant prostate cancer (NCT02415621) use intermittent dosing of abiraterone based on PSA levels, a strategy that can be modeled and adapted [5].
- Simulate Outcomes: Use drug-trial-disease models to simulate the outcomes of different adaptive schedules (e.g., continuous vs. intermittent) before implementing them in the clinic [48].
Quantitative Data and Model Parameters
Table 1: Example Population PK (PopPK) Model Applications in MIPD
| Drug Category | Drug Example | Patient Population | Key Covariate | Dosing Recommendation |
|---|---|---|---|---|
| Antibiotics [46] | Meropenem | Critically ill adults | Renal clearance | Prolonged infusion or high-dosage regimen for patients with high renal clearance. |
| Antiepileptics [46] | Levetiracetam | Critically ill, augmented renal function | Creatinine clearance | Specific dosing schemes proposed based on renal function. |
| Monoclonal Antibodies [46] | Infliximab | Inflammatory bowel disease | Interindividual variability | Dose adaptation; 10 mg/kg improved endoscopic improvement in ulcerative colitis. |
| Tyrosine Kinase Inhibitors [46] | Erlotinib, Imatinib | Cancer patients | Food, drug interactions | Routine TDM recommended due to high PK variability. |
Table 2: Selected Clinical Trials Utilizing Mathematical Models for Treatment Scheduling
| Trial ID | Model/Trial Focus | Cancer Type | Intervention | Status (as of 2025) |
|---|---|---|---|---|
| NCT02595320 [5] | Norton-Simon Model | Metastatic Breast & GI Cancers | Capecitabine (optimized schedule) | Phase 2 (200 patients) |
| NCT02415621 [5] | Adaptive Therapy | Metastatic Prostate Cancer | Intermittent Abiraterone | Early Phase 1 |
| NCT03543969 [5] | Adaptive Therapy | BRAF Mutant Melanoma | Adaptive BRAF-MEK Inhibitor Therapy | Early Phase 1 |
| NCT04388839 [5] | Extinction Therapy | Rhabdomyosarcoma | Evolutionary Therapy | Phase 2, Recruiting |
| NCT05393791 [5] | Adaptive Therapy | Metastatic Prostate Cancer | Adaptive vs. Continuous Abiraterone | Phase 2, Recruiting |
Experimental Protocols for Key Methodologies
Protocol 1: Developing and Qualifying a PopPK Model for MIPD
- Data Collection: Collect rich or sparse pharmacokinetic (PK) samples from the target patient population. Record key patient demographics and clinical data (e.g., weight, renal/hepatic function, concomitant medications) [46].
- Model Development: Use non-linear mixed-effects modeling software (e.g., NONMEM, Monolix) to develop the model. Start with a structural model (e.g., one- or two-compartment) and then identify significant covariates that explain inter-individual variability [46] [48].
- Model Validation: Validate the model using techniques like bootstrap analysis and visual predictive checks. Evaluate its predictive performance internally and, if possible, with an external dataset [48].
- Define Context of Use: Clearly state how the model will be used to inform regulatory decision-making, such as selecting a dose regimen or informing a clinical trial design [48].
- Risk Assessment: Perform a model risk assessment for regulatory submission, considering the "model influence" and "decision consequence" [48].
Protocol 2: Implementing a Bayesian Forecasting Algorithm for Dose Individualization
- Select a Prior Model: Choose a qualified popPK model to serve as the prior.
- Develop an Algorithm: Implement a Bayesian estimation algorithm (e.g., using
Posologyr) that can incorporate one or more drug concentration measurements from an individual patient [46]. - Update and Predict: The algorithm will use the patient's data to update the population prior and derive a posterior parameter set specific to that individual.
- Dose Optimization: Use the individualized parameter set to simulate various dosing regimens and select the one that maximizes the probability of reaching the target exposure (e.g., a specific AUC or trough concentration) [46].
Visualizing Workflows and Relationships
Model-Informed Dosing Workflow
Adaptive Therapy Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Model-Informed Dosing Research
| Tool / Resource | Category | Function / Application | Example / Note |
|---|---|---|---|
| NONMEM | Software | Industry-standard for non-linear mixed effects modeling (popPK/PD). | Used for primary model development [46]. |
| Monolix | Software | Alternative platform for non-linear mixed-effects modeling. | Used for parameter estimation [46]. |
R with Posologyr |
Software | Open-source package for Bayesian dose individualization. | Useful for implementing MIPD in clinical settings [46]. |
| Physiologically-Based Pharmacokinetic (PBPK) Models | Modeling Approach | Mechanistic models to predict PK in virtual populations. | Discussed in FDA MIDD program for predictive safety [48]. |
| Linezolid PopPK Model | Pre-Built Model | Algorithm for dosing optimization in drug-resistant tuberculosis. | An example of a developed MIPD algorithm [46]. |
| FDA MIDD Meeting Program | Regulatory Resource | Pathway for discussing MIPD approaches with regulators. | For dose selection, trial simulation, safety evaluation [48]. |
Overcoming Resistance and Toxicity: Model-Based Solutions for Clinical Hurdles
Addressing Stromal-Induced and Therapy-Induced Resistance
FAQs: Mechanisms and Concepts
What are the main mechanisms of stromal-induced drug resistance? Stromal cells in the tumor microenvironment, particularly Cancer-Associated Fibroblasts (CAFs), promote resistance through several mechanisms. They secrete soluble factors like growth factors and cytokines, remodel the extracellular matrix, reprogram tumor cell metabolism, induce epigenetic modifications in cancer cells, and deliver exosomes. These actions activate pro-survival signaling pathways in cancer cells, protecting them from therapy [49].
How does therapy itself induce resistance? Therapy-induced resistance often occurs through tumor cell plasticity, a reversible phenomenon where cancer cells change their phenotype to evade treatment. This includes processes like Epithelial-Mesenchymal Transition (EMT), transdifferentiation, and the acquisition of a stem-like state. These changes can be driven by non-genetic mechanisms such as epigenetic modifications and activation of key signaling pathways, leading to a population of drug-tolerant persister (DTP) cells [50] [51].
Can stromal-induced resistance be modeled mathematically? Yes, mathematical models using ordinary differential equations can describe the dynamic interactions between cancer cells (C), stromal cells (S), drug concentration (D), and stromal-secreted growth factors (G). These models help identify critical drug concentration thresholds and optimize dosing schedules. The cancer cell growth rate, for instance, can be modeled as a function of drug and growth factor concentration, revealing how the presence of stroma modulates the therapeutic window [52].
Troubleshooting Guides
Problem: Observed Drug Resistance in Co-culture Models
Potential Cause 1: Activation of pro-survival signaling. Soluble factors like HGF or IL-6 from stromal cells activate pathways such as PI3K/Akt and JAK/STAT in cancer cells [49].
- Solution: Implement combination therapy targeting both the primary drug target and the stromal-derived resistance pathway. For example, with EGFR inhibitors, consider adding a c-Met inhibitor if HGF is implicated [49].
- Validation: Perform phospho-protein assays (e.g., Western blot for p-Akt, p-STAT3) on co-cultured cancer cells to confirm pathway activation and subsequent inhibition.
Potential Cause 2: Adhesion-mediated drug resistance. Direct contact between cancer cells and stromal cells via adhesion molecules (e.g., integrin β1) can activate survival signals [53] [54].
- Solution: In experimental models, use blocking antibodies against adhesion molecules like integrin β1 to disrupt contact. For therapeutic intervention, explore agents that target the implicated adhesion signaling.
- Validation: Use flow cytometry to measure surface expression of integrin β1 on cancer cells after co-culture. Assess drug sensitivity with and without blocking antibodies.
Problem: Tumor Cell Plasticity and Phenotypic Switching
Potential Cause: Therapy-induced EMT or lineage switching. Targeted therapies can actively induce a transition to a more drug-tolerant state, such as a mesenchymal or stem-like state [50] [51].
- Solution: Employ intermittent dosing schedules ("drug holidays") to allow DTP cells to revert to a drug-sensitive state. Alternatively, use combination therapies that target the plasticity process itself, such as inhibitors of key transcription factors or epigenetic regulators.
- Validation: Monitor established markers of EMT (e.g., loss of E-cadherin, gain of vimentin) or stemness (e.g., CD44, CD133) in residual cells following treatment.
Experimental Protocols for Key assays
Protocol 1: Assessing Stromal-Induced Resistance in Vitro
Objective: To quantify the protective effect of stromal cells on cancer cell viability during drug treatment. Materials:
- Cancer cells of interest (e.g., colorectal cancer cells)
- Stromal cells (e.g., primary Cancer-Associated Fibroblasts)
- Standard cell culture equipment and reagents
- The therapeutic drug (e.g., cetuximab for CRC)
- Cell viability assay kit (e.g., MTT, CellTiter-Glo)
Method:
- Co-culture Setup: Seed cancer cells and stromal cells in appropriate ratios (e.g., 1:1) in multi-well plates. Include monocultures of cancer cells as controls.
- Drug Treatment: After cells adhere, treat co-cultures and monocultures with a range of drug concentrations. Include a vehicle control (e.g., DMSO).
- Incubation: Incubate for a predetermined period (e.g., 72 hours) to allow for stromal-mediated effects.
- Viability Assay: Quantify cancer cell viability using your chosen assay. For non-contact co-cultures, use transwell systems; for contact co-cultures, you may need to use fluorescently labeled cancer cells and flow cytometry for specific quantification.
- Data Analysis: Calculate the percentage of viable cells for each condition. Compare the IC50 values (drug concentration that kills 50% of cells) between monoculture and co-culture to determine the fold-change in resistance [52] [49].
Protocol 2: Quantifying Signaling Factors from Stromal Cells
Objective: To measure the secretion of resistance-imparting factors from stromal cells in response to therapy. Materials:
- Conditioned media from stromal cell cultures (with and without drug treatment)
- ELISA kits for target factors (e.g., HGF, IL-6, SDF-1)
Method:
- Generate Conditioned Media: Culture stromal cells until 70-80% confluent. Treat with a clinically relevant dose of the drug. After 24-48 hours, collect the media and centrifuge to remove cells and debris.
- ELISA: Perform the enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions for your target factor(s) using the conditioned media.
- Analysis: Compare the concentration of the secreted factor in media from drug-treated versus untreated stromal cells. This identifies therapy-induced changes in the stromal secretome [49].
Mathematical Modeling for Dose Optimization
Mathematical models are crucial for integrating biological data and predicting optimal therapeutic strategies. A core model for stromal-induced resistance can be built using this system of Ordinary Differential Equations (ODEs) [52]:
The cancer growth rate r_C is a function of drug (D) and growth factor (G), often modeled with a Hill function:
Where Dâ‚…â‚€(G) is the drug concentration for 50% effect, which itself increases with G, modeling the right-shift of the dose-response curve.
Key Model Parameters and Outputs Table: Key Parameters for a Stromal-Induced Resistance Model [52]
| Parameter | Description | Typical Units | Impact on Model |
|---|---|---|---|
r_max |
Max. cancer growth rate | 1/day | Higher value = faster tumor growth |
r_min |
Min. cancer growth rate (under high drug) | 1/day | <0 implies tumor reduction is possible |
Dâ‚…â‚€_max |
Max. half-effective drug concentration | nM | Higher value = baseline resistance |
kâ‚, kâ‚‚ |
Hill coefficients for steepness of response | Dimensionless | Higher value = steeper dose-response |
d_D |
Drug decay rate | 1/day | Higher value = faster drug clearance |
| Output | Description | Clinical Relevance | |
D_crit |
Critical drug concentration to shrink tumor | Defines minimum efficacious dose | |
| Therapeutic Window | Range between minimum efficacy and toxicity | Determines safe and effective dosing |
This model can be used to simulate different dosing regimens (continuous vs. intermittent) and identify thresholds that lead to long-term tumor control, informing the design of combination therapies [52].
Research Reagent Solutions
Table: Essential Reagents for Studying Stromal-Induced Resistance [49] [54]
| Reagent / Tool | Function / Target | Application in Research |
|---|---|---|
| Recombinant HGF | Ligand for c-Met receptor | To stimulate resistance pathways in cancer cells; validate HGF-mediated protection. |
| Neutralizing Anti-HGF Antibody | Binds and inhibits HGF | To block HGF/c-Met signaling in co-culture and assess reversal of resistance. |
| Recombinant IL-6 | Pro-inflammatory cytokine | To activate STAT3 signaling and investigate cytokine-induced resistance. |
| Anti-IL-6R Antibody | Blocks IL-6 receptor | To inhibit IL-6 signaling and test its role in stromal-mediated protection. |
| Integrin β1 Blocking Antibody | Cell adhesion molecule | To disrupt direct cancer-stroma contact and study adhesion-mediated drug resistance. |
| CBP/Catenin Inhibitor | disrupts WNT/β-catenin transcription | To target a WNT/β-catenin-mediated EMT program in ALL-stroma co-cultures [54]. |
| c-Met Inhibitor | Tyrosine kinase inhibitor | For combination therapy to overcome HGF-induced resistance to EGFR inhibitors. |
Signaling Pathway and Experimental Workflow Diagrams
Managing Unique Challenges in Immunotherapy Dosing (e.g., CRS in CAR-T)
Frequently Asked Questions (FAQs)
FAQ 1: What are the primary toxicity challenges associated with CAR-T cell therapy?
The most significant toxicity challenges are Cytokine Release Syndrome (CRS) and Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS) [55]. CRS is an excessive inflammatory response triggered by immune activation, with symptoms ranging from fever and chills to life-threatening hypotension, hypoxia, and multi-organ toxicity [56]. It occurs in a high proportion of patients, with any grade of CRS observed in 37% to 93% of cases depending on the product, and severe (Grade 3/4) CRS occurring in 1% to 23% of cases [57]. ICANS involves neurological side effects such as confusion, seizures, and can progress to coma [55].
FAQ 2: How is Cytokine Release Syndrome (CRS) clinically diagnosed and graded?
CRS diagnosis is based on clinical symptoms, which typically begin with fever, often accompanied by malaise, headache, arthralgia, anorexia, rigors, and fatigue [57]. It can rapidly progress to hypoxia, tachycardia, hypotension, and ultimately shock and organ failure. While there is no single diagnostic lab test, laboratory parameters are crucial for monitoring organ dysfunction. The condition is graded on a scale from 1 (mild) to 5 (fatal). Grade 3 is a prolonged reaction not rapidly responding to initial treatments, and Grade 4 involves life-threatening consequences requiring interventions like vasopressors or mechanical ventilation [56]. A key differential diagnosis is infection, so an infectious workup is essential [57].
FAQ 3: What are the current standard management protocols for CRS?
Current management is based on grading [57]:
- Grade 1: Can be managed conservatively on a regular ward with supportive care like fluids and antipyretics.
- Grade 2 and Above: Requires intensive monitoring and intervention. Admission to an Intensive Care Unit (ICU) should be considered starting from Grade 2.
- Anti-Cytokine Therapy: Tocilizumab (an anti-IL-6 receptor antibody) is the FDA and EMA-approved standard for treatment. Prophylactic or preemptive use may reduce the risk of severe CRS [57].
- Steroids: Short courses of steroids (e.g., dexamethasone) can be used and do not seem to have detrimental effects on CAR-T cell efficacy, contrary to earlier beliefs [57].
- Supportive Care: Includes fluids, antipyretics, and vasopressors (which automatically signify higher-grade CRS). Antibiotics are often administered due to the difficulty in ruling out infection [57].
FAQ 4: What strategies are being developed to prevent or mitigate CRS?
Innovative strategies are focused on prevention and better control of CAR-T cell activity [58] [55] [56]:
- Next-Generation CAR-T Engineering: Incorporating Boolean logic gates (e.g., AND, OR gates) can increase specificity, minimizing off-target effects and controlling toxicity. Engineering CAR-T cells with suicide genes (e.g., iCaspase9) allows for their precise elimination if toxicity becomes severe [58].
- Prophylactic Pharmacological Intervention: New oral small molecules are in development to prevent CRS. Examples include CTO1681 (an oral immune modulator in Phase Ib/IIa trials) and POLB 001 (an oral p38 MAPK inhibitor that prevents cytokine production, now Phase II-ready) [55] [56].
- Mathematical Modeling for Dosing: Using models of the cancer-immunity cycle to synchronize chemotherapy and immunotherapy dosing schedules can optimize timing and reduce toxicity. Research suggests that optimized timing can make up for lower total doses, leading to less toxic regimens [59].
FAQ 5: How can mathematical modeling contribute to optimizing immunotherapy dosing in combinations?
Mathematical modeling provides a framework to rationally design combination therapy schedules, moving beyond empirical dose-finding [59] [60]:
- Synchronization with Cancer-Immunity Cycle: Models can test hypotheses about the combination and timing of chemo- and immunotherapy pulses. A key finding is that synchronizing doses with the fundamental period of the cancer-immunity cycle can enhance efficacy and reduce toxicity [59].
- Defining Drug Sequencing: Modeling has shown that immunotherapy dosing should precede chemotherapy dosing and last twice as long for optimal effect in some scenarios [59].
- Understanding Tumor-Immune Dynamics: Fractional calculus models capture the "memory" and history-dependent behavior of tumor-immune interactions, allowing for more realistic simulations and better controller design for adaptive treatment strategies [60].
Troubleshooting Guides
Guide 1: Troubleshooting Severe CRS in Preclinical CAR-T Models
This guide addresses the challenge of severe, uncontrollable CRS in animal models, which can halt development.
| Step | Problem/Symptom | Possible Cause | Recommended Solution & Experimental Protocol |
|---|---|---|---|
| 1 | Rapid onset of severe hypothermia, hypotension, and mortality post CAR-T infusion. | Overly potent CAR-T activation; excessive cytokine production; too high tumor burden or CAR-T cell dose. | Implement a "Suicide Gene" Safety Switch. Experimental Protocol: Co-transduce your CAR construct with an inducible suicide gene (e.g., iCaspase9). Administer the dimerizing drug (AP1903 for iCaspase9) at the first sign of severe toxicity (e.g., sustained hypothermia) and monitor for rapid ablation of CAR-T cells and stabilization of vital signs [58]. |
| 2 | High levels of pro-inflammatory cytokines (IL-6, IFN-γ, TNF-α) and severe CRS, but loss of antitumor efficacy with corticosteroid use. | Broad immunosuppression from steroids blunting CAR-T cell function. | Use Targeted Cytokine Blockade Prophylactically. Experimental Protocol: Administer an anti-IL-6R antibody (e.g., tocilizumab) or a JAK inhibitor prior to the onset of severe CRS symptoms. Compare cytokine levels, CRS scores, and tumor volume measurements against a control group receiving only rescue therapy [57]. |
| 3 | "On-target, off-tumor" toxicity leading to CRS-like symptoms from damage to healthy tissues. | CAR-T target antigen is expressed at low levels on healthy cells. | Incorporate Logic Gates into CAR Design. Experimental Protocol: Develop a CAR-T system requiring two antigens for full activation (e.g., an AND-gate CAR). In your model, demonstrate that cytotoxicity and cytokine release are robust only in dual-antigen positive tumor cells, while dual-antigen negative healthy cells are spared [58]. |
Guide 2: Troubleshooting Inefficient Combination Therapy Dosing Schedules
This guide addresses the problem of suboptimal efficacy and increased toxicity when combining immunotherapy with chemotherapy.
| Step | Problem/Symptom | Possible Cause | Recommended Solution & Experimental Protocol |
|---|---|---|---|
| 1 | Combination therapy shows no improvement, or even antagonism, compared to monotherapy. | Drug scheduling is negating the immune-stimulating effects of either agent. | Synchronize Dosing with the Cancer-Immunity Cycle. Experimental Protocol: Using a mathematical model of your system, identify the fundamental period of the tumor-immune cycle. Test a regimen where a pulse of immunotherapy precedes a chemotherapy pulse. For example, model and then validate in vivo that immunotherapy given for half a cycle, followed by chemotherapy for a quarter cycle, yields superior results [59]. |
| 2 | Excessive toxicity when combining therapies, limiting the usable dose. | Overlapping toxicities and maximum tolerated dose (MTD) is exceeded. | Apply Model-Informed Drug Development (MIDD) for Dose Optimization. Experimental Protocol: Leverage preclinical PK/PD and toxicity data to build a quantitative model. Use this model to simulate various dose combinations and schedules to identify a "therapeutic window" where efficacy is maintained but toxicity is minimized. Prioritize these regimens for in vivo testing, focusing on lower, pulsed doses rather than continuous MTD [19]. |
| 3 | High patient-to-patient variability in response to a fixed combination schedule. | The one-size-fits-all schedule does not account for individual dynamic differences. | Utilize Fractional Calculus for Personalized Scheduling. Experimental Protocol: Employ a fractional-order dynamical model (e.g., using Caputo or Atangana-Baleanu operators) to fit individual patient tumor-immune time-series data. The fractional order can capture patient-specific "memory" and dynamics. Use model forecasts to personalize the timing and duration of therapy pulses for each patient [60]. |
Quantitative Data Tables
Table 1: Key Cytokines in CRS and Their Roles as Therapeutic Targets
Table detailing the primary cytokines involved in Cytokine Release Syndrome, their functions, and associated targeting agents.
| Cytokine | Primary Cell Source | Role in CRS Pathogenesis | Targeted Therapy (Examples) |
|---|---|---|---|
| IL-6 | Macrophages, T cells | A key driver of systemic inflammation; induces fever, activates acute phase response [56]. | Tocilizumab (anti-IL-6R), Siltuximab (anti-IL-6) [57]. |
| TNF-α | Macrophages, T cells | Promotes inflammation, endothelial activation, and contributes to vascular leak and hypotension [56]. | POLB 001 (p38 MAPK inhibitor - prevents synthesis) [56]. |
| IFN-γ | CAR-T cells, NK cells | Activates macrophages, enhances antigen presentation, and contributes to CRS severity [58]. | JAK inhibitors (indirectly, by blocking signaling) [56]. |
| IL-1 | Macrophages | Pyrogen, promotes inflammation and tissue damage. | Anakinra (IL-1 receptor antagonist) [57]. |
Table 2: Clinical Grading and Management of Cytokine Release Syndrome (CRS)
A structured overview of the clinical presentation and recommended interventions for different grades of CRS, based on current guidelines [57] [56].
| CRS Grade | Clinical Presentation | Recommended Management & Monitoring |
|---|---|---|
| Grade 1 | Fever, possibly with malaise, headache, arthralgia. | Supportive care (antipyretics, fluids). Monitor on regular ward. |
| Grade 2 | Fever with hypotension responsive to fluids; hypoxia requiring low-flow oxygen (<40%). | Moderate intervention. Consider ICU transfer. Administer Tocilizumab. Supportive care. |
| Grade 3 | Fever with hypotension requiring vasopressors; hypoxia requiring high-flow oxygen (≥40%). | Aggressive intervention. Admit to ICU. Administer Tocilizumab and Steroids (e.g., Dexamethasone). |
| Grade 4 | Life-threatening; requiring mechanical ventilation or significant organ support. | Intensive life support. Maximal intervention with Tocilizumab and high-dose steroids. |
| Grade 5 | Death. | - |
Research Reagent Solutions
Table of key reagents and technologies for investigating and mitigating CRS in immunotherapy research.
| Research Reagent / Technology | Primary Function | Application in CRS Management Research |
|---|---|---|
| Boolean Logic Gate CARs | Engineered CAR-T cells that require multiple antigens for full activation (e.g., AND-gate). | Increases tumor specificity, reduces "on-target, off-tumor" toxicity, and minimizes aberrant activation that leads to CRS [58]. |
| Inducible Suicide Genes (iCaspase9) | Safety switch that allows for ablation of CAR-T cells upon administration of a small molecule drug. | Used as a fail-safe mechanism to rapidly eliminate CAR-T cells in case of severe, uncontrollable CRS [58]. |
| p38 MAPK Inhibitors (e.g., POLB 001) | Oral small molecule that inhibits p38 MAPK, a key regulator of cytokine production. | Investigated as a prophylactic agent to prevent the release of cytokines like TNF-α and IL-6, thereby reducing the incidence and severity of CRS [56]. |
| JAK/STAT Inhibitors (e.g., Ruxolitinib) | Small molecule that blocks signaling downstream of multiple cytokine receptors. | Used to mitigate CRS by blunting the cellular response to a wide array of cytokines; caution is needed as it may also impair antitumor efficacy [56]. |
| Anti-IL-6R Antibody (Tocilizumab) | Monoclonal antibody that blocks the interleukin-6 receptor. | The clinical standard for treatment of moderate to severe CRS; used in both rescue and preemptive settings in research to define optimal management protocols [57]. |
Experimental Workflows and Signaling Pathways
CAR-T CRS Signaling Pathway
Combination Therapy Optimization
Prophylactic CRS Inhibition
Navigating Narrow Therapeutic Windows with Adaptive Therapy Strategies
What is a Narrow Therapeutic Window, and why is it a central challenge in oncology drug development?
A narrow therapeutic window refers to a small dosage range where a drug is effective without causing unacceptable toxicity. In oncology, this is a particularly critical challenge because many cancer drugs, including targeted therapies, have a minimal difference between the dose required for efficacy and the dose that causes severe side effects. Historically, the standard approach has been to use the maximum tolerated dose (MTD), determined through short-term toxicity studies. However, research indicates this often leads to poorly optimized treatments; for instance, nearly 50% of patients on late-stage trials for small molecule targeted therapies require dose reductions, and the FDA has required additional dosing studies for over 50% of recently approved cancer drugs [1].
How do Adaptive Therapy strategies fundamentally differ from the standard Maximum Tolerated Dose approach?
Adaptive Therapy (AT) represents a paradigm shift from the static MTD model. Instead of continuously administering the highest possible dose to try and eradicate a tumor, AT uses dynamic, patient-specific dosing schedules. The core principle is to leverage intra-tumoral competition between drug-sensitive and drug-resistant cancer cell populations [61]. Treatment is applied intermittently—often at the MTD when active—but is withdrawn or reduced to allow a controlled population of drug-sensitive cells to survive and suppress the growth of resistant clones through competition for resources. This approach aims for long-term tumor control rather than immediate eradication, significantly extending the time to disease progression compared to continuous therapy [61].
What role does Mathematical Modeling play in optimizing these adaptive strategies?
Mathematical modeling provides the quantitative framework necessary to design and personalize adaptive therapy protocols. It moves beyond trial-and-error by using computational models to simulate tumor dynamics and predict how different dosing schedules will affect cancer growth and evolution. Key roles include:
- Informing Protocol Design: Models identify optimal triggers for treatment application and withdrawal. For example, a "threshold-based" protocol (AT-N*) administers treatment only when the tumor size exceeds a pre-specified, patient-specific threshold, which modeling has shown can significantly increase time to progression [61].
- Integrating Complex Data: Model-Informed Drug Development (MIDD) uses approaches like Quantitative Systems Pharmacology (QSP) and exposure-response modeling to integrate non-clinical and clinical data. This helps understand the relationship between drug exposure, preliminary activity, and adverse reactions, supporting the selection of optimized dosing regimens [62] [22].
- Accounting for Clinical Realities: Models can be tailored to incorporate practical constraints, such as the discrete time intervals between patient monitoring appointments, ensuring that derived protocols are clinically feasible [61].
FAQs on Implementation and Workflow
What are the primary types of mathematical models used in this field?
The following table summarizes the key computational tools used in Model-Informed Drug Development for oncology.
| Model/Methodology | Primary Function and Application |
|---|---|
| Quantitative Systems Pharmacology (QSP) | Incorporates biological mechanisms to understand and predict a drug's therapeutic and adverse effects with limited clinical data. Useful for developing dosing strategies to reduce the risk of specific adverse reactions [22]. |
| Exposure-Response (ER) Modeling | Analyzes the relationship between drug exposure (e.g., concentration in the body) and its effectiveness or safety. Used to predict the probability of efficacy and adverse reactions for dosing regimens not directly tested in trials [62] [22]. |
| Population Pharmacokinetic (PPK) Modeling | Describes the pharmacokinetics and inter-individual variability in a patient population. Can be used to select dosing regimens that achieve target exposure and to transition from weight-based to fixed dosing [22]. |
| Fractional-Order Models | An advanced approach that captures "memory effects," providing a more accurate representation of biological processes with long-term dependencies, such as cancer progression and treatment response [63]. |
| Lotka-Volterra & Competitive Dynamics Models | Ordinary differential equation models that explicitly describe the competition between drug-sensitive and drug-resistant cancer cell populations, forming the basis for many adaptive therapy simulation frameworks [61]. |
How do I design a first-in-human (FIH) trial that supports future adaptive therapy development?
Moving beyond the traditional "3+3" dose-escalation design is crucial. To design a FIH trial that generates data useful for adaptive therapy planning, consider these methodologies [1]:
- Use Model-Informed Starting Doses: Employ mathematical models that consider factors beyond simple weight-based allometric scaling from animals, such as receptor occupancy rates, to determine safer and potentially more effective starting doses.
- Implement Novel Escalation Designs: Utilize model-informed dose-escalation designs that respond to both efficacy measures and late-onset toxicities, offering more nuanced decision-making than algorithmic approaches.
- Incorporate Backfill and Expansion Cohorts: Increase the number of patients treated at certain dose levels of interest within the early-stage trial. This generates more robust clinical data on the benefit-risk ratio at those doses.
- Collect Biomarker Data: Integrate biomarker testing (e.g., measuring changes in circulating tumor DNA) to help identify tumor responses that may not be detected with short follow-up times.
Our team is planning a registrational trial. What model-informed approaches can support final dosage selection?
For the final dosage decision, the FDA encourages a holistic approach that utilizes the totality of efficacy and safety data. Successful model-informed approaches include [22] [1]:
- Clinical Utility Index (CUI): A quantitative framework that collaboratively integrates diverse data (efficacy, safety, pharmacokinetics) to determine the most promising dose(s).
- Logistic Regression of Safety Data: A safety-focused model that analyzes key landmark safety data (e.g., incidence of dose interruptions or reductions) across different dosages from early trials to model the probability of adverse reactions.
- Seamless Adaptive Trial Designs: These combine traditionally distinct trial phases (e.g., dose-finding and confirmatory) into a single study. This allows for more rapid enrollment, faster decision-making, and the accumulation of more long-term safety and efficacy data to better inform the final dosing decision.
- Leveraging Models for Combination Therapies: For combination therapies, such as an mRNA-based cancer vaccine and an anti-CTLA-4 antibody, modeling can optimize the regimen. For instance, a study found that the antibody should be administered following vaccination, with its dose positively correlating with the vaccine dose within a safe range [36].
Adaptive Therapy Clinical Workflow
Troubleshooting Common Scenarios
Problem: Our model suggests an adaptive protocol, but it requires continuous tumor monitoring, which is not clinically feasible.
Solution: Develop protocols that account for discrete monitoring intervals. A key advancement in making adaptive therapy clinically practical is deriving optimal treatment thresholds that acknowledge patients are only seen at specific appointments (e.g., every 30 days) [61]. Instead of a protocol that requires immediate action the moment a tumor size threshold is crossed, design a "threshold-based" strategy (AT-N) where treatment decisions are made only at these discrete visits. The model itself can be used to determine the optimal threshold size (N^{}) that maximizes time to progression given the specific monitoring interval (\tau) [61].
Problem: We observe high heterogeneity in patient responses to the same adaptive protocol in our simulations.
Solution: Personalize the protocol parameters using patient-specific data. Heterogeneity in outcomes is expected because tumor dynamics vary significantly between patients [61]. The solution is to move from a one-size-fits-all adaptive protocol to a personalized one. Use the mathematical modeling framework to calibrate model parameters (e.g., growth rates of sensitive and resistant cells) to individual patient data, such as initial tumor size and early response kinetics. This allows for the derivation of a patient-specific optimal treatment threshold (N^{*}) or a time-varying threshold that adapts to changes in the patient's tumor dynamics over the course of therapy [61].
Problem: We are developing a combination therapy (e.g., immunotherapy + targeted therapy) and are unsure how to sequence and dose the agents to minimize side effects.
Solution: Use a fractional-order model integrated with feedback control. For complex combination regimens, a fractional-order model can better capture the memory effects and hereditary traits of biological systems [63]. Integrate this with a Proportional-Integral-Derivative (PID) controller, a feedback mechanism that dynamically adjusts drug dosages based on the difference between the desired (e.g., target tumor size) and actual state. This creates a dynamic and adaptive treatment strategy that optimizes the combination, sequence, and timing of therapies to control the tumor while minimizing side effects [63].
Model-Informed Protocol Development
The Scientist's Toolkit: Research Reagent Solutions
The following table details key resources and computational tools essential for research in this field.
| Tool / Resource | Function in Research |
|---|---|
| Model-Informed Drug Development (MIDD) | A framework that employs quantitative models to support drug development and regulatory decision-making, from discovery to post-market surveillance [62]. |
| Virtual Population Simulation | A computational technique that creates realistic virtual patient cohorts to predict pharmacological and clinical outcomes under varying conditions and dosing regimens [62]. |
| Clinical Trial Simulation Software | Software that uses mathematical models to virtually predict trial outcomes and optimize study designs before conducting actual clinical trials [62]. |
| Fit-for-Purpose Initiative (FDA) | A regulatory pathway that provides a framework for the regulatory acceptance of dynamic tools, including models, for use in specific contexts in drug development [22] [1]. |
| Bayesian Inference & Gaussian Processes | Statistical methods for quantifying uncertainty in model parameters and guiding optimal experimental design, especially useful with limited datasets [64]. |
| Prostate-Specific Antigen (PSA) | A widely accepted biomarker for tumor burden in prostate cancer, enabling regular, non-invasive monitoring essential for implementing adaptive therapy protocols [61]. |
| Quantitative Systems Pharmacology (QSP) Models | Integrative models that combine systems biology and pharmacology to generate mechanism-based predictions on drug behavior and treatment effects [62] [22]. |
Utilizing Clinical Utility Indices for Holistic Benefit-Risk Analysis
Frequently Asked Questions (FAQs) on Clinical Utility Indices
1. What is a Clinical Utility Index (CUI), and what is its primary purpose in drug development? A Clinical Utility Index (CUI) is a quantitative, multi-attribute decision-making tool used to integrate and weigh multiple efficacy and safety outcomes into a single composite score [65]. Its primary purpose is to support benefit-risk assessment when multiple attributes are involved in a decision, helping to understand the relevance of each attribute and differentiate compounds from competitors [65]. It provides a transparent and collaborative mechanism to integrate data and determine concrete doses of interest, moving beyond decisions based solely on short-term toxicity [1].
2. When in the drug development process is it most beneficial to use a CUI? The use of a CUI is most beneficial during early clinical development to support early-stage decision-making, such as selecting doses for further exploration in a proof-of-concept trial [1] [65]. It is particularly useful before the final dosage decision for the large registrational trial [1]. A probabilistic CUI is recommended for early decisions due to its practicality, reasonable accuracy, and transparency, at stages where financial factors are less critical [65].
3. What are the common challenges when constructing a CUI, and how can they be mitigated? Common challenges include:
- Endpoint Selection and Weighting: Determining which efficacy and safety endpoints to include and assigning appropriate weights to reflect their clinical importance can be challenging. Methods to address this involve collaboration with clinicians, statisticians, and patient advocates [66], and using techniques like multi-criteria decision analysis [65].
- Double Counting and Correlated Endpoints: There is a risk of over-emphasizing an outcome if correlated endpoints are included. Careful evaluation of the relationships between different endpoints is required [65].
- Handling Uncertainty: Early clinical data often has uncertainty. Using a probabilistic CUI model can help characterize this uncertainty [65].
4. How does a CUI support the optimization of combination therapies? A CUI provides a structured framework to evaluate the complex benefit-risk profile of multiple drugs used together. For example, a multicriteria decision analysis model has been applied to estimate the benefit-risk of a combination therapy for overactive bladder, underlining the benefit of a quantitative approach in clinical development programs [65]. By combining utility functions for efficacy and toxicity, an overall multiattribute utility function can be developed for a treatment, which is essential for complex combination regimens [65].
5. What is the relationship between a CUI and Model-Informed Drug Development (MIDD) approaches? CUI is a key component within the broader MIDD paradigm. While population pharmacokinetic-pharmacodynamic (PK/PD) and exposure-response models predict drug concentrations and responses, the CUI provides the framework to synthesize these predictions into a holistic benefit-risk score [22] [1]. Frameworks like CUI can be part of the amalgamation of data that provides a rationale for dosage selection, often informed by model-generated data [1].
Troubleshooting Guides
Issue 1: Difficulty in Selecting and Weighting Endpoints for the CUI
Problem: Researchers struggle to choose the most relevant efficacy and safety endpoints and to assign appropriate weights that reflect their relative importance in the overall benefit-risk assessment.
Solution:
- Stakeholder Engagement: Collaborate with a multidisciplinary team, including clinicians, statisticians, and patient advocates, to refine the study design and ensure all critical perspectives are captured [66].
- Structured Prioritization: Employ formal multi-criteria decision analysis (MCDA) techniques to systematically evaluate and weight endpoints [65].
- Leverage Preliminary Data: Use all available nonclinical and early clinical data to inform the initial selection. This can include pharmacodynamic biomarker data, preliminary overall response rates, and incidence of adverse events from early trials [22].
Issue 2: The CUI Fails to Discriminate Between Dosing Regimens
Problem: The calculated CUI values for different doses are similar, making it difficult to identify an optimal dose.
Solution:
- Refine Endpoint Sensitivity: Re-evaluate the chosen endpoints. Incorporate more sensitive measures, such as changes in circulating tumor DNA (ctDNA) levels, which can help identify responses not detected with traditional endpoints due to short follow-up [1].
- Expand Data Collection: Utilize backfill and expansion cohorts in early-stage trials. These cohorts increase the number of patients at specific dose levels of interest, providing more robust clinical information to strengthen the understanding of the benefit-risk ratio [1].
- Incorporate Time-Dynamics: The basic CUI is often a landmark analysis. Consider implementing a longitudinal model that accounts for the time to and duration of events, such as time to first dosage modification or duration of toxicity [22].
Issue 3: Integrating CUI with Complex Mathematical Models
Problem: Researchers find it challenging to connect the output of mechanistic PK/PD or tumor growth models to the inputs required for the CUI.
Solution:
- Define Linking Framework: Establish a clear workflow where mathematical models provide predicted values for key efficacy and safety endpoints. These predicted values then serve as the quantitative inputs for the CUI calculation.
- Use Simulation: Leverage model-based simulations to predict the probability of adverse reactions and tumor response as a function of drug exposure [22]. These simulated outcomes can then be fed into the CUI framework to compare the potential benefit-risk of possible dosing regimens virtually [22].
- Adopt a Fit-for-Purpose Approach: Tailor the complexity of the CUI to the available data and the stage of development. A simpler model might be used initially, with complexity increasing as more data becomes available [1].
Experimental Protocols & Data Presentation
Protocol for Implementing a CUI in an Early-Phase Oncology Trial
This protocol outlines the methodology for using a CUI to select doses for further exploration based on preliminary activity and safety data [1].
1. Objective To quantitatively compare multiple dosage regimens of a new oncology drug using a CUI that integrates key efficacy and safety data, thereby identifying an optimized dosage for evaluation in a subsequent proof-of-concept trial.
2. Pre-Trial Requirements
- Data Sources: Gather all relevant nonclinical and clinical data, including pharmacokinetics, pharmacodynamics, preliminary efficacy (e.g., overall response rate), and safety (e.g., grade 3+ adverse events, incidence of dose interruptions/reductions) [22].
- Stakeholder Alignment: Convene a meeting with clinicians, statisticians, clinical pharmacologists, and patient advocates to agree on the endpoints and their weights for the CUI [66].
3. Step-by-Step Methodology
- Step 1: Attribute Selection. Define the efficacy and safety attributes to include in the index. Example attributes are shown in Table 1.
- Step 2: Utility Function Definition. For each attribute, define a utility function that transforms the raw data (e.g., incidence of an AE, ORR) onto a standardized scale (e.g., 0 to 1), where 1 is most desirable.
- Step 3: Weight Assignment. Assign a weight to each attribute to reflect its relative importance in the overall benefit-risk assessment. The sum of all weights should equal 1.
- Step 4: CUI Calculation. For each dosage regimen under consideration, calculate the CUI using the following formula:
CUI = (Weight₠× Utilityâ‚) + (Weightâ‚‚ × Utilityâ‚‚) + ... + (Weightâ‚™ × Utilityâ‚™) - Step 5: Analysis and Decision. Compare the CUI values across different dosages. The dose with the highest CUI represents the most favorable balance of benefit and risk. Sensitivity analysis should be performed to test the robustness of the conclusion against changes in the weights.
Quantitative Data Tables
Table 1: Example Efficacy and Safety Attributes for a CUI in Oncology
| Data Area | Attribute | Description / Measurement | Utility Function Direction |
|---|---|---|---|
| Clinical Efficacy | Overall Response Rate (ORR) | Proportion of patients with tumor shrinkage of a predefined amount [22]. | Higher is better |
| Clinical Efficacy | Effect on Surrogate Biomarker | e.g., log change in circulating tumor DNA (ctDNA) levels [1]. | Higher is better |
| Clinical Safety | Incidence of Grade 3+ Adverse Events | Proportion of patients experiencing severe side effects [22]. | Lower is better |
| Clinical Safety | Incidence of Dose Reduction | Proportion of patients requiring a dose reduction due to toxicity [22]. | Lower is better |
| Patient Reported Outcomes | Quality of Life Score | Score from a validated questionnaire (e.g., EORTC QLQ-C30) [22]. | Higher is better |
Table 2: Example CUI Calculation for Three Hypothetical Doses
| Dose Level | ORR Utility (W=0.5) | Gr3+ AE Utility (W=0.3) | Dose Reduction Utility (W=0.2) | CUI Score |
|---|---|---|---|---|
| Dose A (High) | 0.9 | 0.4 | 0.5 | 0.71 |
| Dose B (Medium) | 0.8 | 0.8 | 0.8 | 0.80 |
| Dose C (Low) | 0.5 | 1.0 | 1.0 | 0.75 |
In this example, Dose B has the highest CUI, indicating a better balance of efficacy and tolerability.
Workflow Visualization
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CUI Implementation
| Item / Solution | Function in CUI Analysis |
|---|---|
| Clinical Data Management System (CDMS) | A secure database platform for collecting, storing, and managing structured clinical trial data, including efficacy endpoints, adverse events, and patient-reported outcomes, which serve as the raw inputs for the CUI. |
| Statistical Analysis Software (e.g., R, SAS) | Software used to perform the statistical calculations for the CUI, including generating utility functions, applying weights, computing final scores, and conducting sensitivity analyses. |
| Multi-Criteria Decision Analysis (MCDA) Framework | A structured methodological framework (which may be implemented in software or as a set of guidelines) that provides the formal process for selecting, weighting, and combining multiple criteria into a single index like the CUI. |
| Model-Informed Drug Development (MIDD) Tools | Software for population PK/PD modeling, exposure-response analysis, and tumor growth modeling. These tools generate predictive data that can be used as inputs for the CUI, especially for doses not directly tested. |
From Virtual to Reality: Validating Models in Clinical Trials and Regulatory Frameworks
Model-Informed Drug Development (MIDD) is an essential framework for advancing drug development and supporting regulatory decision-making. MIDD plays a pivotal role in drug discovery and development by providing quantitative predictions and data-driven insights that accelerate hypothesis testing, enable more efficient assessment of potential drug candidates, reduce costly late-stage failures, and ultimately accelerate market access for patients [62]. Evidence from drug development and regulatory approval has demonstrated that a well-implemented MIDD approach can significantly shorten development cycle timelines, reduce discovery and trial costs, and improve quantitative risk estimates, particularly when facing development uncertainties [62].
The application of MIDD has been particularly transformative in the context of combination therapies, where mathematical models help unravel complex drug-drug interactions and optimize dosing regimens for multiple agents administered simultaneously. For clinical trial professionals, MIDD provides a structured approach to address critical questions such as: "Which models will provide the best insights for this indication at this stage?" and "How can we optimize clinical trial design including dosage optimization?" [62]. The International Council for Harmonization (ICH) has further standardized MIDD practices across different countries and regions through expanded guidance, including the M15 general guidance, promoting more consistent application of MIDD in global drug development and regulatory interactions [62].
Table 1: Key MIDD Approaches in Clinical Development
| Modeling Approach | Primary Application in Clinical Trials | Relevant Trial Phase |
|---|---|---|
| Physiologically Based Pharmacokinetic (PBPK) | Predicting drug-drug interactions, organ impairment effects | Phase 1-3 |
| Population PK/PD (PPK/ER) | Characterizing variability in drug exposure and response | Phase 1-3 |
| Quantitative Systems Pharmacology (QSP) | Mechanism-based prediction of treatment effects and side effects | Phase 1-2 |
| Model-Based Meta-Analysis (MBMA) | Contextualizing trial results against existing compounds | Phase 2-3 |
| Clinical Trial Simulation | Optimizing study designs and predicting outcomes | Phase 2-3 |
| Bayesian Inference | Integrating prior knowledge with observed data for improved predictions | Phase 1-3 |
Success Stories: Model-Informed Trials in Oncology
Optimizing Checkpoint Inhibitor Combination Therapy
A recent groundbreaking application of MIDD in oncology involved optimizing combination therapy with immune checkpoint inhibitors. Researchers implemented a Quantitative Systems Pharmacology (QSP) model that integrated known biological pathways of T-cell activation, tumor proliferation, and drug mechanism of action to identify optimal dosing schedules for combination immunotherapy [62]. The model successfully predicted that staggered dosing of two immunotherapies would yield superior efficacy compared to concurrent administration, a finding subsequently validated in a Phase 2 clinical trial.
The QSP framework incorporated tumor growth dynamics, immune cell infiltration, and receptor occupancy to simulate clinical outcomes across different dosing regimens. This approach allowed researchers to virtually test multiple combination scenarios, significantly reducing the number of patients required to identify optimal dosing in actual clinical trials [62]. The trial design incorporated model-informed biomarkers and endpoint selection, demonstrating a 30% improvement in objective response rate compared to standard dosing approaches.
Table 2: Key Research Reagents for QSP in Immuno-Oncology
| Research Reagent/Model Component | Function in Experiment |
|---|---|
| QSP Platform Software | Integrates biological pathways and drug properties for mechanism-based predictions |
| Virtual Patient Population | Creates diverse, realistic virtual cohorts to predict outcomes under varying conditions |
| Immune Cell Trafficking Module | Simulates T-cell infiltration into tumor microenvironment |
| Checkpoint Inhibitor PK/PD Model | Characterizes drug exposure and receptor occupancy relationships |
| Tumor Growth Dynamic Model | Describes tumor proliferation and response to immune-mediated killing |
Troubleshooting Guide: Common Issues in QSP Modeling
FAQ: Why might a QSP model fail to predict clinical outcomes accurately, and how can this be addressed?
QSP models may yield inaccurate predictions due to several common issues:
Insufficient Model Calibration: When models are not adequately calibrated to human pathophysiology, predictions may diverge from actual clinical outcomes. Solution: Implement a stepwise calibration process using available clinical data before making prospective predictions [67].
Inadequate Representation of Variability: Failure to account for patient-to-patient variability can limit model utility. Solution: Incorporate virtual population simulations that reflect true biological diversity [62].
Overly Complex Model Structure: Unnecessary complexity without sufficient data for parameter estimation can reduce model reliability. Solution: Apply "fit-for-purpose" principles, ensuring model complexity aligns with the specific question of interest and available data [62].
Systematic troubleshooting should follow these steps: First, repeat the model verification process to ensure computational implementation matches theoretical design. Second, verify that all input parameters are biologically plausible and properly referenced. Third, conduct sensitivity analysis to identify parameters with disproportionate influence on outcomes [68]. Finally, compare model predictions against any available preliminary data, and if discrepancies exist, systematically evaluate each model component [67].
Success Stories: Model-Informed Trials in Metabolic Diseases
Novel Antibody Therapy for Obesity-Related Disorders
Novartis researchers recently applied MIDD approaches to assess the feasibility of a novel antibody therapy for obesity-related disorders targeting the GDF15-GFRAL pathway [69]. The therapeutic concept involved an antibody designed to bind endogenous GDF15 to extend its half-life, thereby enhancing GFRAL signaling to reduce food intake and promote weight loss [69]. The team employed a mechanistic PK/PD model for subcutaneous administration with a structure similar to published models of antibody-ligand traps.
The model incorporated standard monoclonal antibody PK parameters for cynomolgus monkeys and humans, exploring how drug PK, dosing regimen, antibody affinity, and patient variability in baseline GDF15 levels would impact circulating GDF15 concentrations [69]. Simulations demonstrated that sufficiently high total GDF15 concentrations could be achieved to drive meaningful weight loss, informing both Go/No-Go decisions and optimal therapeutic design. The model further explored combination approaches using mixtures of stabilizing therapeutic antibody and recombinant GDF15 at various ratios to optimize exposure profiles [69].
Diagram: GDF15 Antibody Mechanism for Obesity Treatment
Troubleshooting Guide: Common PK/PD Modeling Issues
FAQ: What are common failure points in PK/PD modeling for metabolic diseases, and how can they be resolved?
Common issues in metabolic disease PK/PD modeling include:
Inaccurate Parameter Estimation: When PK parameters are not properly estimated from preclinical species, human predictions may be unreliable. Solution: Use Bayesian inference approaches to integrate prior knowledge with observed data for improved predictions [64].
Failure to Account for Disease Progression: Models that don't incorporate natural disease progression may misattribute effects. Solution: Include disease progression modules calibrated to control arm data [69].
Overlooked Food Effects: For metabolic diseases, food-drug interactions may significantly impact exposure. Solution: Incorporate PBPK elements to simulate food effects on drug absorption [62].
When troubleshooting PK/PD models, researchers should first verify that the structural model appropriately represents the underlying biology. Next, examine residual plots to identify systematic biases. If unexpected variability patterns emerge, consider incorporating additional covariates or adjusting statistical models. Finally, conduct visual predictive checks to assess model performance across the concentration-response range [67] [68].
Table 3: Troubleshooting PK/PD Modeling Problems
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Poor model fit | Structural model misspecification, influential outliers | Evaluate alternative model structures, examine residual plots |
| High unexplained variability | Missing covariates, inadequate dosing records | Incorporate patient-specific factors, verify data quality |
| Failed predictive checks | Overfitting, external factors not accounted for | Simplify model, include additional physiological factors |
| Unstable parameter estimates | Insufficient data, correlated parameters | Utilize Bayesian priors, collect more informative data |
MIDD in Regulatory Submissions and 505(b)(2) Applications
The use of MIDD has become increasingly important in regulatory submissions, particularly for 505(b)(2) applications and generic drug product development [62]. Regulatory agencies now frequently employ modeling and computer simulations at various phases of drug discovery and development, with the FDA's Center for Drug Evaluation and Research (CDER) using these approaches to inform review decisions [70]. Success stories include the use of PBPK models to generate evidence for generic drug product development in bioequivalence studies, an approach referred to as Model-Integrated Evidence (MIE) [62].
In one notable case, a pharmaceutical company used MIDD approaches to support a 505(b)(2) application for a combination therapy, leveraging existing safety data for individual components while using model-informed evidence to support the combination regimen [62]. The application incorporated clinical trial simulations that integrated literature data with limited new clinical data to demonstrate combination efficacy and safety, significantly reducing development costs and time to market. The model-informed approach provided a comprehensive framework for evaluating different dosing scenarios and identifying the optimal risk-benefit profile for the combination therapy.
Implementing MIDD: Technical Protocols and Workflows
Standard Protocol for Model-Informed Trial Design
Implementing a successful model-informed clinical trial requires a systematic approach:
Step 1: Define Context of Use (COU) and Questions of Interest Clearly articulate the specific decisions the model will inform and the key questions it should address. This establishes the "fit-for-purpose" scope for model development [62].
Step 2: Data Collection and Curation Assemble all relevant data, including preclinical PK/PD, in vitro assays, prior clinical data, and literature information. Ensure data quality through rigorous validation procedures [70].
Step 3: Model Selection and Development Select appropriate modeling methodologies based on the COU. Common approaches include PBPK for absorption and drug-interaction predictions, QSP for mechanism-based efficacy assessment, and population PK/PD for variability characterization [62].
Step 4: Model Qualification and Verification Verify that the computational implementation matches theoretical specifications and qualify the model against existing data to establish predictive performance [67].
Step 5: Clinical Trial Simulation Execute virtual trials using the qualified model to explore different design options, dosing regimens, and patient population characteristics [62].
Step 6: Prospective Application and Validation Implement the model-informed design in the actual clinical trial and collect data to validate model predictions [62].
Step 7: Model Refinement Update the model with new clinical data as it becomes available, refining predictions for subsequent trial phases [69].
Diagram: MIDD Implementation Workflow
Essential Research Reagent Solutions
Table 4: Key Reagents and Tools for MIDD Implementation
| Tool Category | Specific Solutions | Application in MIDD |
|---|---|---|
| Modeling Software | NONMEM, Monolix, GastroPlus, Simbiology | PK/PD model development, PBPK modeling, QSP platform |
| Data Management | R, Python, SAS | Data curation, visualization, statistical analysis |
| Clinical Trial Simulators | Trial Simulator, East | Virtual patient generation, study design optimization |
| Visualization Tools | Spotfire, GraphPad Prism | Results communication, exploratory data analysis |
| Database Resources | PubChem, ClinicalTrials.gov, DrugBank | Literature data sourcing, competitive landscape analysis |
Future Directions and Emerging Technologies
The future of model-informed clinical trials is increasingly intertwined with artificial intelligence (AI) and machine learning (ML) approaches [62]. These technologies are enhancing traditional MIDD methods by analyzing large-scale biological, chemical, and clinical datasets to make predictions, recommendations, or decisions that influence real or virtual environments [62]. ML techniques are being employed to enhance drug discovery, predict ADME properties, and optimize dosing strategies based on multidimensional patient characteristics [62].
Emerging applications include the use of AI-driven patient stratification models that identify subgroups most likely to respond to combination therapies, and digital twin technologies that create virtual representations of individual patients to optimize therapeutic strategies. However, these advanced approaches face challenges including lack of appropriate resources, slow organizational acceptance and alignment, and the need for further validation of AI/ML models in regulatory contexts [62]. Despite these challenges, the continued expansion of MIDD promises to further streamline drug development, particularly for complex combination therapies requiring sophisticated optimization approaches.
The integration of MIDD into combination therapy development represents a paradigm shift in clinical research, moving from empirical dose-finding to mechanism-based, quantitative approaches that leverage the totality of available data. As these methodologies continue to evolve and demonstrate success across therapeutic areas, they are poised to become standard practice in clinical development, ultimately accelerating the delivery of innovative therapies to patients.
The primary objective of Phase I oncology trials is to identify a safe and effective dose for further development. For decades, the traditional 3+3 design was the predominant method, used in over 95% of published Phase I oncology trials [71] [72]. However, the mechanisms of action of targeted therapies and immunotherapies have challenged the underlying assumption that the maximum tolerated dose (MTD) constitutes the optimal dose [72] [1]. This has catalyzed a shift toward model-informed dosing approaches, which use mathematical models to integrate complex data and tailor dosing strategies more precisely [73]. This analysis compares these two paradigms, providing a technical resource for scientists optimizing combination therapies.
Understanding the Core Designs
The Traditional 3+3 Design
The 3+3 design is an algorithm-based, rule-driven approach. It involves treating successive cohorts of three patients at increasing dose levels. The decision to escalate, de-escalate, or declare a dose as the MTD is based strictly on the observed number of dose-limiting toxicities (DLTs) within each cohort [74].
- Key Methodology:
- A cohort of 3 patients is treated at a starting dose.
- If 0 of 3 experience a DLT, escalate to the next higher dose for the next cohort.
- If 1 of 3 experiences a DLT, expand the cohort to 6 patients at the same dose.
- If 1 of 6 experiences a DLT, escalation continues. If at least 2 of 6 experience a DLT, the MTD is considered exceeded.
- The MTD is typically defined as the dose level below which at least 2 of 3 or 2 of 6 patients experience a DLT [71].
Model-Informed Dosing Approaches
Model-informed dosing employs statistical and mathematical models to guide dose selection and optimization, using all accumulated data. It encompasses several designs used in early development and Model-Informed Precision Dosing (MIPD), which focuses on tailoring doses for individual patients in clinical practice [46] [45] [73].
Common Model-Informed Designs for Trials:
- CRM (Continual Reassessment Method): A model-based design that continuously updates the probability of toxicity for all dose levels based on all collected data after each patient outcome to determine the best next dose [75].
- BOIN (Bayesian Optimal Interval): A model-assisted design that uses pre-specified toxicity probability intervals to guide escalation and de-escalation, balancing simplicity and statistical rigor [75] [74].
- BLRM (Bayesian Logistic Regression Model): A model-based approach that excels at incorporating historical data and is well-suited for combination therapy studies [75].
Key Methodology of MIPD:
- Develop a population pharmacokinetic (PopPK) and/or pharmacokinetic/pharmacodynamic (PK/PD) model using available data [46] [76].
- Identify a target exposure range associated with both efficacy and safety (e.g., a trough concentration,
Cmin,ss) [76]. - For an individual patient, use Bayesian forecasting to integrate their specific characteristics (e.g., weight, renal function) and drug measurements (e.g., from Therapeutic Drug Monitoring) to estimate a personalized dose that achieves the target exposure [46].
Comparative Analysis: Performance and Characteristics
Quantitative Design Comparison
Table 1: Key Characteristics of Dose-Finding Designs
| Feature | Traditional 3+3 | BOIN (Model-Assisted) | CRM (Model-Based) | MIPD (Clinical Practice) |
|---|---|---|---|---|
| Core Principle | Algorithm-based rules | Pre-specified toxicity intervals | Continuous model reassessment | Bayesian forecasting & PopPK models |
| Statistical Foundation | Limited | Bayesian | Bayesian | Bayesian / Frequentist |
| MTD Identification Accuracy | Low [77] [72] | Higher [75] [74] | High [75] | Not Applicable (Individual-level) |
| Patient Safety | Conservative, but poor safety profile in combination trials [77] | Built-in overdose control [75] | Dependent on model specification | High (aims to optimize individual safety) |
| Implementation Complexity | Low | Moderate [75] | High [75] | High (requires specialized software/expertise) [46] |
| Regulatory Acceptance | Historical standard | Growing recognition [74] | Established, but complex [75] | Supported by FDA initiatives like MIDD [1] |
| Use in Combination Therapies | Poor, often reduces to one-dimensional search [77] | Supported by specific extensions (e.g., BOIN combo) [77] | Supported, but model complexity increases [77] | Applicable, but requires complex drug-drug interaction models |
Performance Metrics in Clinical Trials
Table 2: Comparative Performance in Simulated Trials
| Performance Metric | Traditional 3+3 | Model-Assisted (e.g., BOIN) | Model-Based (e.g., CRM, BLRM) |
|---|---|---|---|
| Probability of Selecting True MTD | Low [77] [72] | Competitive & High [77] [74] | High, but can be variable [75] [77] |
| Risk of Overdosing (Patients > MTD) | Variable, can be high in combinations [77] | Competitive and balanced safety profile [77] | Can be high in some designs, dependent on safety rules [77] |
| Trial Duration | Long due to delays [71] | Shorter (efficient allocation) | Shorter (efficient allocation) |
| Handling Delayed Outcomes | Poor; requires suspension or ad-hoc rules [71] | Good; with specific extensions [74] | Good; with specific extensions |
Technical Guide: Implementing Model-Informed Approaches
Experimental Protocol for a Model-Informed Combination Trial
Objective: To identify the Maximum Tolerated Dose (MTD) contour for a two-drug combination (Drug A and Drug B) using the BOIN combination design.
Materials & Software:
- Statistical Software: R with
BOINpackage or equivalent commercial software. - Pre-clinical Data: Prior information on single-agent toxicity and potential drug-drug interactions.
- Protocol Parameters: Pre-specified target toxicity rate (e.g., 30%), dose levels for each drug, cohort size (typically 3), and sample size.
Methodology:
- Design Phase:
- Specify the matrix of dose combinations to be investigated.
- Simulate thousands of trials under different toxicity scenarios to characterize the design's operating characteristics (OC), including probability of correct MTD selection and patient safety.
- Finalize and document the trial design and decision rules in the protocol.
- Trial Execution Phase:
- Collar the first cohort of patients at the lowest dose combination (Drug A L1, Drug B L1).
- Observe DLTs: After the pre-defined DLT assessment window (e.g., 4 weeks), record the number of DLTs for the cohort.
- Apply BOIN decision rules:
- Calculate the observed DLT rate at the current dose combination.
- Compare this rate to the pre-calculated escalation/de-escalation boundaries (λe, λd).
- The next cohort is assigned to a dose combination based on this comparison. The BOIN algorithm allows for flexible escalation/de-escalation across the two-dimensional grid [77] [74].
- Continue enrolling cohorts until a pre-specified sample size is reached or the trial is stopped for safety.
- MTD Selection: At trial end, use model averaging or an isotonic regression estimator applied to all collected data to identify the MTD(s) or MTD contour [74].
Workflow for Model-Informed Precision Dosing (MIPD)
The following diagram illustrates the cyclic process of implementing MIPD in a clinical setting.
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Resources for Model-Informed Drug Development
| Tool / Reagent | Function / Application | Example Use Case |
|---|---|---|
| NONMEM | Industry-standard software for population PK/PD model development. | Building a PopPK model for a new monoclonal antibody to explain variability in drug exposure [46]. |
R / Python with specialized packages (e.g., BOIN, dfcrm) |
Open-source platforms for implementing adaptive trial designs and statistical analysis. | Simulating the operating characteristics of a BOIN combination trial before protocol finalization [77] [74]. |
| PBPK (Physiologically-Based Pharmacokinetic) Software (e.g., GastroPlus, Simcyp) | Simulates drug absorption, distribution, metabolism, and excretion based on physiology. | Predicting the likelihood of a drug-drug interaction between two combination agents prior to clinical testing. |
| Validated Bioanalytical Assay (e.g., LC-MS/MS) | Precisely measures drug concentrations in biological matrices (plasma, serum). | Generating the PK data required for PopPK model development and for TDM in MIPD [76]. |
| Clinical Data Collector Tools | Aggregates real-world patient data from electronic health records for model refinement. | Extracting pazopanib concentrations and liver enzyme levels for a real-world exposure-toxicity analysis [76]. |
Frequently Asked Questions (FAQs)
Q1: The 3+3 design is simple and understood by everyone. Why should I adopt a more complex model-informed approach? A: While simple, the 3+3 design is inefficient and inaccurate. It fails to use all available data, leading to a lower probability of correctly identifying the true MTD. It is particularly unsuitable for combination therapy trials and the development of modern targeted therapies, where the goal is often to find an optimal biological dose rather than just the MTD [77] [72] [1]. Model-informed designs offer superior accuracy and efficiency, which is a key reason behind regulatory initiatives like FDA's Project Optimus [1].
Q2: How do I handle delayed dose-limiting toxicities (DLTs) in a model-informed trial, which is a common issue in immunotherapy? A: Standard model-informed designs like CRM and BOIN have time-to-event extensions (TITE-CRM, TITE-BOIN) that account for this. These methods incorporate the actual follow-up time for patients who have not yet completed the DLT assessment window, weighting their data appropriately. This allows for continuous patient enrollment without suspending the trial, significantly reducing study duration [71] [74].
Q3: What is the difference between model-based designs used in Phase I trials and Model-Informed Precision Dosing (MIPD) used in clinical practice? A: The primary difference is the goal. Model-based trial designs (e.g., CRM, BLRM) aim to find a population-level recommended dose (e.g., MTD) for a new drug. In contrast, MIPD is applied after a dose is approved to optimize the dose for an individual patient in clinical practice, using their specific characteristics and drug concentration measurements to achieve a target exposure [46] [73] [76].
Q4: Our team has limited statistical expertise. Can we still implement a model-informed design like BOIN? A: Yes. Model-assisted designs like BOIN were created to bridge this gap. They have pre-calculated decision boundaries that can be tabulated in the study protocol, making them almost as easy to implement as the 3+3 design but with much better statistical properties. User-friendly software and apps are also available to facilitate their implementation [75] [74].
Q5: For a drug with a known exposure-response relationship, how is MIPD operationalized? A: The process, as demonstrated with pazopanib, involves:
- Defining a therapeutic target window (e.g., trough concentration
Cmin,ssof 20.5-34 mg/L for efficacy and avoiding liver toxicity) [76]. - Using a validated PopPK model to estimate the dose that will achieve this target for an individual patient based on their covariates.
- Initiating treatment with this dose, then measuring the actual drug concentration (TDM).
- Using Bayesian feedback to refine the PK parameter estimates and adjust the dose until the patient's exposure is within the target window [46] [76].
The Role of Digital Twins and Virtual Patient Cohorts in De-risking Development
Frequently Asked Questions (FAQs) and Troubleshooting Guides
This technical support resource provides practical guidance for researchers using digital twins and virtual patient cohorts to de-risk the development of combination therapies.
Fundamentals of Digital Twins
What is a Digital Twin in healthcare, and how does it differ from a standard computational model? A Digital Twin (DT) is an integrated, data-driven virtual representation of a real-world patient or population. It is characterized by a dynamic, bidirectional flow of information between the physical entity and its digital counterpart [78] [79]. Unlike static computational models, a true DT is individualized, interconnected, interactive, informative, and impactful (the 5Is) [78]. It continuously updates with real-time data from its physical twin, enabling predictive simulation and decision support [80] [81]. For therapy development, this means you can test interventions on the virtual model before administering them to a real patient.
What are the main types of Digital Twins, and which is most relevant for preclinical drug development? The evolution of DTs can be understood through a maturity model [78]:
- Static Twin: A digital replica with only static properties.
- Mirror/Functional Twin: A static twin with dynamic behavior capabilities, often used for surgical planning or in-silico clinical trial design.
- Shadow/Self-adaptive Twin: A functional twin that can acquire real-time data and update itself, used for medical device design and biomarker discovery.
- Intelligent Twin: A self-adaptive twin with autonomous learning, reasoning, and acting capabilities, enabling personalized medicine and wellness management. For preclinical development, Mirror and Shadow Twins are most immediately applicable for simulating trial designs and predicting patient responses to combination therapies [82] [78].
Mathematical Modeling and In-Silico Trials
How can virtual patient cohorts address the challenges of generalizability and recruitment in traditional clinical trials? Traditional Randomized Clinical Trials (RCTs) often have restrictive eligibility criteria, leading to systematically under-represented demographic and clinical groups. This limits the external validity of the results and makes patient recruitment slow and expensive [82]. Virtual patient cohorts, generated via AI and deep generative models, replicate the underlying structure and variability of real-world populations [82]. They can be used in two key ways [82]:
- As Synthetic Controls: Each real participant is paired with a digital twin whose disease progression is projected under standard care, providing comparator data without exposing additional patients to a placebo.
- As Virtual Treatment Groups: Virtual patients receive the experimental therapy in silico, allowing researchers to explore efficacy and safety signals before human administration. This approach can reduce the required sample size for real-world trials, shorten timelines, and lower costs while improving the diversity of the tested population [82] [83].
We are building a mathematical model to optimize the scheduling of a combination therapy (e.g., TRT and CAR-T cells). Our model parameters are not converging. What should we check? Parameter instability in mechanistic models for combination therapy often stems from issues with model structure or data fitting. Follow this troubleshooting guide:
| Problem Area | Specific Checks | Potential Solutions |
|---|---|---|
| Model Identifiability | Check for parameters with high correlation. Assess if available data is sufficient to estimate all parameters. | Simplify the model by fixing less sensitive parameters. Perform a sensitivity analysis (e.g., using Sobol indices) to identify key drivers. Increase the diversity of data used for calibration (e.g., include monotherapy and combination data with different timings) [84]. |
| Data Integration | Verify the quality and scale of experimental data used for calibration. Ensure units are consistent. | Use global optimization algorithms (e.g., particle swarm, genetic algorithms) to avoid local minima. Incorporate data from multiple sources (e.g., pharmacokinetics, tumor burden, immune cell counts) to better constrain the model [84] [85]. |
| Numerical Implementation | Check the Ordinary Differential Equation (ODE) solver for stability. Verify that initial conditions are realistic. | Reduce solver step size or try a different solver algorithm (e.g., from Runge-Kutta to Adams/BDF). Test a range of physiologically plausible initial conditions. |
Our model for a CAR-T and Targeted Radionuclide Therapy (TRT) combination in Multiple Myeloma is not capturing the antagonistic effect we see experimentally when therapies are administered too close together. What component might be missing?
Your model may be missing the critical element of radiation-induced killing of the therapeutic cells. The validated mathematical framework for TRT and CAR-T cell therapy includes a specific term for the radiation dose rate (kRxi) affecting both tumor cells (NT) and CAR-T cells (NC) [84]. The system of differential equations is:
dNT/dt = ÏNT - H(t-Ï„_TRT) kRx_T NT - H(t-Ï„_CART) k1 NT NCdNC/dt = k2 (NT+NR) NC - H(t-Ï„_TRT) kRx_C NC - θ NC
Ensure your model includes a component, like the kRx_C term, that accounts for the radiation sensitivity (αC) of the CAR-T cells. Without this, the model cannot predict the antagonism that occurs when active CAR-T cells are exposed to TRT [84].
Implementation and Validation
What are the key steps to building a patient-specific digital twin for combination therapy optimization? A practical, step-by-step protocol for creating a foundational DT is as follows [86] [81]:
- Gather Structured Biomarker Data: Collect comprehensive baseline clinical information, including symptoms, lab results, imaging data, genetic profiles, and omics data (genomics, proteomics) from EHRs and disease registries.
- Integrate Wearable and Lifestyle Data: Incorporate continuous data streams from wearable devices (e.g., heart rate, activity) and lifestyle factors to create a dynamic view of the patient's status.
- Centralize and Unify Patient Information: Create a single, unified data platform that integrates all disparate data sources into a "single source of truth."
- Develop and Calibrate the Mathematical Model: Build a mechanistic model of the disease and therapy interaction. Use the collected data to calibrate and validate the model parameters. For combination therapies, this often involves differential equations for tumor-immune-drug interactions [84] [85].
- Run Simulations and Predict Outcomes: Use the calibrated DT to simulate various combination therapy scenarios, testing different doses, sequences, and timings to identify an optimal regimen.
- Validate and Refine with Real-World Evidence: Continuously update the DT with new patient data and validate its predictions against observed outcomes to iteratively improve its accuracy.
We have developed a digital twin for a medical device. What is required for regulatory submission to bodies like the FDA? Regulatory bodies like the FDA now provide guidance for submissions involving computational modeling and simulation [83] [87]. A robust credibility assessment framework is essential. Your submission should demonstrate [83]:
- Verification: Confirmation that the computational model is implemented correctly and without error. This involves checking the code and numerical calculations.
- Validation: Evidence that the model's predictions accurately represent real-world physiological and clinical outcomes. This requires comparing model predictions against independent experimental or clinical data.
- Uncertainty Quantification: Analysis of the uncertainty in model predictions, stemming from both parameter variability and model form limitations. Engage with regulatory experts early to draft a virtual testing plan connected to your physical testing. The FDA's ASME V&V 40 standard provides a framework for assessing credibility based on the model's risk and context of use [87].
Experimental Protocols and Workflows
Workflow for a Virtual Clinical Trial
The following diagram illustrates a generalized workflow for designing and executing a virtual clinical trial using digital twins, synthesized from multiple research applications [82] [83] [87].
Virtual Clinical Trial Workflow
Mathematical Modeling for Combination Therapy
This diagram outlines the key components and logical flow of a mechanistic mathematical model used to optimize the dosing and scheduling of combination therapies, as demonstrated in oncology research [84] [85].
Combination Therapy Modeling Logic
Quantitative Data and Research Reagents
Performance Metrics of Digital Twins in Healthcare
The table below summarizes quantitative data on the performance of Digital Twins across various clinical applications, as reported in recent literature [81].
| Clinical Application | Reported Performance Metric | Quantitative Result | Context / Model Used |
|---|---|---|---|
| Cardiology | Reduction in AF recurrence rate | 40.9% vs 54.1% (control) | Patient-specific cardiac DT for drug selection [81]. |
| Cardiology | ECG monitoring classification | 85.77% Accuracy, 95.53% Precision | CardioTwin architecture for real-time monitoring [81]. |
| Neurology (Parkinson's) | Disease prediction accuracy | 97.95% Accuracy | DT-based remote healthcare system [81]. |
| Neurology (Brain Tumor) | Radiotherapy planning | 16.7% dose reduction | Personalized planning for high-grade gliomas [81]. |
| Metabolic (Type 1 Diabetes) | Time in target glucose range during exercise | Increased from 80.2% to 92.3% | Exercise Decision Support System (exDSS) [81]. |
| Metabolic (Type 1 Diabetes) | Hypoglycemia incidents during aerobic exercise | Reduced from 15.1% to 5.1% | Exercise Decision Support System (exDSS) [81]. |
| Oncology (Lung Cancer) | Clinical variable forecasting (R²) | 0.98 | DT-GPT model [81]. |
| Oncology (Chest X-ray) | Image classification | 96.8% Accuracy, 92% Precision | Lung-DT framework with YOLOv8 [81]. |
Key Research Reagent Solutions for Mathematical Modeling
This table details essential "reagents" – in this context, key data types and computational tools – required for building and calibrating mathematical models of combination therapies [84] [86] [85].
| Research Reagent / Data Type | Function in Modeling & Experimentation | Example Sources |
|---|---|---|
| Preclinical Monotherapy Data | Provides baseline for calibrating model parameters for each therapeutic agent independently before modeling their combination. Critical for verifying individual agent effects. | In-vivo animal studies; historical control data from previous trials [84]. |
| Longitudinal Tumor Burden Data | Serves as the primary outcome measure for calibrating tumor cell dynamics (proliferation rate Ï) and validating model predictions against experimental results. | Caliper measurements; medical imaging (MRI, CT) [84]. |
| Therapeutic Cell Counts (e.g., CAR-T) | Used to calibrate parameters for immune cell dynamics, including proliferation/exhaustion (k₂) and clearance (θ) rates within the model. | Flow cytometry; blood samples [84]. |
| Radiobiological Parameters (α, β) | Define the sensitivity of tumor and healthy cells to radiation therapy. These are central to the Linear-Quadratic model used in TRT simulations. | Literature from radiobiology; cell survival curve assays [84]. |
| Physiological & Anatomical Data | Informs the initial conditions and constraints of the model, ensuring virtual patients or tumors are physiologically plausible. | EHRs; medical imaging; wearable device data [86] [81]. |
| Validated Computational Solver | A robust numerical solver for integrating the system of differential equations that constitute the mechanistic model over time. | ODE solvers (e.g., in MATLAB, R, or Python with SciPy) [84]. |
| Global Optimization Algorithm | Used for model calibration to find the set of parameters that best fits the experimental data, helping to avoid local minima. | Particle Swarm Optimization; Genetic Algorithms [85]. |
Regulatory Pathways and Fit-for-Purpose Model Submissions
Frequently Asked Questions (FAQs) and Troubleshooting Guides
FAQ 1: What does "fit-for-purpose" mean in the context of a model submission to the FDA?
A fit-for-purpose (FFP) model is one that is strategically aligned with a specific "Question of Interest" and "Context of Use" (COU) for a given stage of drug development. [62]
- What it is: A FFP model is tailored to answer a specific scientific or clinical question (e.g., "What is the recommended starting dose for a First-in-Human trial?") within a clearly defined context. The model's complexity, data inputs, and validation should be appropriate for this intended use. [62]
- What it is not: A model is not FFP when it fails to define the COU, uses poor quality or insufficient data, or lacks proper verification and validation. Oversimplification or unjustified complexity can also render a model not FFP. [62] For example, a machine learning model trained on one clinical scenario may not be FFP for predicting outcomes in a different setting. [62]
FAQ 2: We are developing two novel investigational drugs as a combination therapy. What are the key regulatory expectations for demonstrating the "contribution of effect" (COE) for each drug?
The FDA's draft guidance on cancer drug combinations emphasizes the need to characterize how each drug contributes to the overall treatment benefit. [88] While full factorial trials (which include monotherapy arms) are often preferred, the agency acknowledges they are not always feasible. [89]
- Troubleshooting Guide: Challenges with Factorial Designs
- Challenge: Factorial trials require large sample sizes, making them impractical for rare cancers or biomarker-defined populations. [89]
- Solution: The FDA is open to alternative approaches when justified. You should provide a strong biological rationale, such as synthetic lethality or compelling biomarker-driven data, for omitting monotherapy arms. [89] Engage with the agency early to discuss alternative designs like adaptive trials or the use of external controls. [89]
- Challenge: A drug may have limited monotherapy activity but show compelling effects in combination.
- Solution: Use Model-Informed Drug Development (MIDD) principles, such as Quantitative Systems Pharmacology (QSP) or exposure-response models, to build evidence for the contribution of each component. [89]
FAQ 3: What alternative data sources can be used to support a model submission when clinical data is limited?
Regulators are increasingly accepting diverse data sources to support evidence generation.
- Accepted Sources: You may use Real-World Data/Evidence from electronic health records, claims data, and registries. [89] Additionally, data from other histologies (with the same biomarker), information extrapolated from a different treatment line, or MIDD approaches can be leveraged. [89]
- Key Consideration: When using external data, you must explicitly address comparability. This includes assessing patient-level RWD, biomarker status, and other clinically relevant covariates to ensure the data is appropriate for your defined COU. [89]
FAQ 4: Our model-informed drug development (MIDD) approach suggests a different optimal dose than what was identified by our traditional 3+3 dose escalation trial. How should we proceed?
This is a common issue, as the traditional 3+3 design focuses primarily on short-term toxicity and often identifies a Maximum Tolerated Dose that may not be the most efficacious or best-tolerated dose for longer-term treatment. [1]
- Recommended Action: Follow the principles of FDA's Project Optimus. Conduct a dose-optimization study that directly compares multiple doses (including the one identified by your model) to fully assess the benefit-risk profile. [1]
- Methodology: Employ population pharmacokinetic-pharmacodynamic (PopPK/PD) and exposure-response (ER) modeling on your clinical data. These techniques can help identify optimized dosages by combining safety and efficacy evaluations and can extrapolate the effects of doses not clinically tested. [1]
FAQ 5: What are the common reasons for a "fit-for-purpose" model to be rejected during a regulatory submission?
A model submission can face challenges for several key reasons:
- Poorly Defined Context of Use: The model's purpose is not clearly articulated or is too vague. [62]
- Inadequate Validation: The model lacks proper verification, calibration, or validation for its stated COU. [62]
- Data Quality Issues: The model is built on data of insufficient quality or quantity to support its conclusions. [62]
- Lack of Regulatory Engagement: Failure to seek early feedback from regulators on the modeling strategy, especially when using novel approaches or alternative trial designs. [89] [1]
Experimental Protocols for Key Modeling Approaches
Protocol 1: Developing a Quantitative Systems Pharmacology (QSP) Model for Combination Therapy Dose Optimization
Objective: To develop a mechanistic QSP model that simulates the interaction between two drugs in a combination therapy and predicts their synergistic effect on a clinical endpoint.
Methodology:
- Systems Biology Model Construction: Develop a mathematical model representing the key biological pathways targeted by each drug. This typically involves ordinary differential equations (ODEs) describing:
- Tumor cell proliferation and death.
- Target receptor occupancy and downstream signaling for each drug.
- Immune cell activation and tumor infiltration (if applicable).
- Crosstalk mechanisms between the two drug pathways. [40]
- Model Calibration: Parameterize the model using in vitro co-culture data and in vivo animal model data for each drug alone and in combination. Fit the model to time-course data on tumor volume and relevant biomarker levels. [40]
- Virtual Population Simulation: Generate a diverse virtual patient population by varying key system parameters (e.g., receptor expression levels, metabolic rates) within physiologically plausible ranges. [62]
- Clinical Trial Simulation: Simulate a virtual clinical trial by administering different dose levels and schedules of the combination therapy to the virtual population. Predict outcomes such as progression-free survival (PFS) and the incidence of key adverse events. [62]
- Dose-Optimization Analysis: Identify the dose combination that maximizes a clinical utility index (CUI), which quantitatively balances predicted efficacy and safety. [1]
Protocol 2: Implementing a Model-Informed First-in-Human (FIH) Dose Algorithm
Objective: To determine the safe and biologically active starting dose for a novel investigational drug using a model-based approach, moving beyond allometric scaling.
Methodology:
- Data Collection: Gather preclinical data including in vitro IC50 (half-maximal inhibitory concentration), in vivo PK data from animal models, and receptor occupancy/ binding affinity data. [1]
- PBPK/PD Model Development: Construct a Physiologically Based Pharmacokinetic-Pharmacodynamic (PBPK/PD) model. This model incorporates human physiology, drug-specific properties, and the mechanism of action to predict human PK and PD. [62]
- Prediction of Human Exposure: Use the PBPK model to simulate human PK profiles and predict the exposure (e.g., AUC, Cmax) expected at various dose levels. [62]
- Identification of Biologically Active Dose: Determine the dose required to achieve exposures predicted to result in a target level of receptor occupancy or pathway modulation, based on the PD model. [1]
- Safety Margin Assessment: Compare the predicted human exposure at the biologically active dose to exposures associated with toxicity in animal models to establish a safety margin. [1]
- FIH Dose Recommendation: Recommend a starting dose that is expected to be both safe and to show pharmacological activity, which may be higher than doses derived from traditional safety-based calculations alone. [1]
Research Reagent Solutions: Essential Materials for Model Development
The table below details key computational and data resources essential for building regulatory-grade fit-for-purpose models.
| Item Name | Function/Explanation |
|---|---|
| Quantitative Systems Pharmacology (QSP) Platform | An integrative modeling framework that combines systems biology with pharmacology to generate mechanism-based predictions on drug behavior and treatment effects. [62] |
| PBPK Modeling Software | Software used for Mechanistic PBPK modeling to understand the interplay between human physiology and drug properties, critical for predicting human PK and FIH doses. [62] |
| Population PK/PD Analysis Tool | Software for performing Population PK (PPK) and Exposure-Response (ER) analysis to understand variability in drug exposure and its relationship to effectiveness or adverse effects. [62] |
| Clinical Trial Simulator | A tool that uses mathematical and computational models to virtually predict trial outcomes and optimize study designs before conducting actual trials. [62] |
| Model-Based Meta-Analysis (MBMA) | A technique that integrates summary-level data from multiple clinical trials to quantify drug treatment effects and disease progression, providing context for new drug development. [62] |
| Virtual Population Generator | A computational technique that creates diverse, realistic virtual cohorts of individuals to predict pharmacological or clinical outcomes under varying conditions. [62] |
Visualizations of Signaling Pathways and Workflows
Diagram 1: Model Submission Workflow
Diagram 2: CAR-T Cell Signaling for QSP
Diagram 3: Model Validation Framework
Conclusion
Mathematical modeling represents a fundamental shift in oncology drug development, moving the field beyond the one-size-fits-all maximum tolerated dose toward dynamic, personalized combination therapy optimization. By integrating foundational biological principles with advanced computational methods, these models provide a powerful tool to decipher complex tumor dynamics, predict and overcome resistance, and design optimal dosing strategies that maximize efficacy while minimizing toxicity. The successful application of these approaches in clinical trials for various cancers, supported by regulatory initiatives like Project Optimus, underscores their immense translational potential. Future directions will involve tighter integration with AI and real-world data, the expansion of virtual patient frameworks, and tackling the complexities of long-term therapy management. For researchers and drug developers, embracing these model-informed strategies is no longer optional but essential for creating the next generation of smarter, more effective cancer treatments.
References
- 1. Key Considerations for Improving Dosage Optimization in ... [https://www.aacr.org/blog/2025/10/03/key-considerations-for-improving-dosage-optimization-in-oncology/]
- 2. Review Dose optimization in cancer drug development [https://www.sciencedirect.com/science/article/pii/S0959804925003752]
- 3. A mathematical framework for comparison of intermittent ... [https://www.nature.com/articles/s41540-024-00461-2]
- 4. Cancer is complex and dynamic—ergo, dose optimization ... [https://cancerletter.com/trials-and-tribulations/20220513_7/]
- 5. Mathematical Oncology: How Modeling Is Transforming ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12614879/]
- 6. New Report Supports Overhaul of Cancer Drug Dosing [https://www.asco.org/about-asco/press-center/news-releases/new-report-supports-overhaul-cancer-drug-dosing]
- 7. Dosing Threeâ€Drug Combinations That Include Targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5423521/]
- 8. Learning from failed model predictions [http://mathematical-oncology.org/blog/learning-from-failed-model-predictions.html]
- 9. Optimal dosage protocols for mathematical models of ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1303814/full]
- 10. Overview of the epigenetic/cytotoxic dual-target inhibitors ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523424011176]
- 11. Mathematical Modelling for Optimal Vaccine Dose Finding [https://pmc.ncbi.nlm.nih.gov/articles/PMC9144167/]
- 12. Introduction to Mathematical Oncology - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6752950/]
- 13. Dose–response relationship - Wikipedia [https://en.wikipedia.org/wiki/Dose%E2%80%93response_relationship]
- 14. The Effect of Ongoing Exposure Dynamics in Dose Response Relationships | PLOS Computational Biology [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000399]
- 15. Dose Response Modeling - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/dose-response-modeling]
- 16. Tumour heterogeneity and resistance to cancer therapies [https://www.nature.com/articles/nrclinonc.2017.166]
- 17. Eco-evolutionary dynamics [https://en.wikipedia.org/wiki/Eco-evolutionary_dynamics]
- 18. Combination Therapy Dose Optimization in Oncology Trials [https://www.iqvia.com/library/white-papers/combination-therapy-dose-optimization-in-oncology-trials]
- 19. Oncology Combination Drug Development Strategies for ... [https://pubmed.ncbi.nlm.nih.gov/39365679/]
- 20. Mathematical Modeling, Computational Methods ... [https://www.nitmb.org/mathematical-modeling]
- 21. Realizing the promise of Project Optimus: Challenges and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11098159/]
- 22. FDA-AACR Strategies for Optimizing Dosages for Oncology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12536354/]
- 23. Project Optimus, an FDA initiative: Considerations for ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2023.1144056/full]
- 24. FDA's Project Optimus: What Pharma and Biotech Need to ... [https://www.precisionformedicine.com/blog/fdas-project-optimus-what-pharma-and-biotech-need-to-know]
- 25. Agent-based vs. equation-based multi-scale modeling for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10810539/]
- 26. Integrated pharmacokinetic-pharmacodynamic and agent ... [https://www.sciencedirect.com/science/article/abs/pii/S0169409X24002965]
- 27. Pharmacokinetic ODE models in Stan - Notes from a data witch [https://blog.djnavarro.net/posts/2023-05-16_stan-ode/]
- 28. Mechanistic data-informed multiscale quantitative systems ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12516987/]
- 29. Multiscale co-simulation design pattern for neuroscience ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10895016/]
- 30. Neural-ODE for pharmacokinetics modeling and its advantage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8283337/]
- 31. A beginner's guide to AnyLogic model errors [https://www.anylogic.com/blog/a-beginner-s-guide-to-anylogic-model-errors/]
- 32. 7 Common Myths About Quantitative Systems Pharmacology [https://www.allucent.com/resources/blog/common-misconceptions-about-qsp-modeling]
- 33. How Do We “Validate†a QSP Model? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6157666/]
- 34. Quantitative systems modeling approaches towards ... [https://www.frontiersin.org/journals/systems-biology/articles/10.3389/fsysb.2022.1063308/full]
- 35. Methodologies for Quantitative Systems Pharmacology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5572127/]
- 36. Mathematical Modelling and Optimization of Medication ... [https://pubmed.ncbi.nlm.nih.gov/40455115/]
- 37. QSP Software [https://www.simulations-plus.com/software/qsp-software/]
- 38. Modelling and simulation: questions and answers [https://www.ema.europa.eu/en/human-regulatory-overview/research-and-development/scientific-guidelines/clinical-pharmacology-pharmacokinetics/modelling-simulation-questions-answers]
- 39. In silico study of heterogeneous tumour-derived organoid ... [https://www.nature.com/articles/s41598-024-63125-5]
- 40. Mathematical models and computational approaches in ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1581210/full]
- 41. Computational model of CAR T-cell immunotherapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9730379/]
- 42. A Mathematical Modeling Approach for Targeted ... [https://www.mdpi.com/2072-6694/13/20/5171]
- 43. In silico assessment of cellular damage from Luâ€177, Ac†... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12559466/]
- 44. Mathematical Modeling Unveils Optimization Strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11142146/]
- 45. Model-informed precision dosing: State of the art and future ... [https://www.sciencedirect.com/science/article/pii/S0169409X24002436]
- 46. Model-Informed Precision Dosing (MIPD) - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9780803/]
- 47. Mathematical modeling of combined therapies for treating ... [https://www.sciencedirect.com/science/article/pii/S0025556424000300]
- 48. Model-Informed Drug Development Paired Meeting Program [https://www.fda.gov/drugs/development-resources/model-informed-drug-development-paired-meeting-program]
- 49. The Role of Tumor-Stroma Interactions in Drug Resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8173135/]
- 50. Tumor cell plasticity in targeted therapy-induced resistance [https://www.nature.com/articles/s41392-023-01383-x]
- 51. Tumor cell plasticity in targeted therapy-induced resistance [https://pubmed.ncbi.nlm.nih.gov/36906600/]
- 52. Modeling critical dosing strategies for stromal-induced ... [https://www.nature.com/articles/s41540-025-00495-0]
- 53. Cancer-stromal interactions: Role in cell survival ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3230306/]
- 54. Stromal-induced epithelial-mesenchymal transition ... [https://www.sciencedirect.com/science/article/pii/S221112472300815X]
- 55. CAR T's biggest hurdle: solving the toxicity problem [https://www.drugtargetreview.com/article/186787/car-ts-biggest-hurdle-solving-the-toxicity-problem/]
- 56. CRS prevention: Taking the sting from CAR-T's tail [https://www.ddw-online.com/crs-prevention-taking-the-sting-from-car-ts-tail-34667-202504/]
- 57. Management of Cytokine Release Syndrome (CRS) and HLH [https://www.ncbi.nlm.nih.gov/books/NBK584171/]
- 58. Challenges and innovations in CAR-T cell therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC11196618/]
- 59. Gaming the cancer-immunity cycle by synchronizing ... [https://pubmed.ncbi.nlm.nih.gov/40779253/]
- 60. Mathematical modeling of tumor-immune dynamics [https://www.nature.com/articles/s41598-025-13683-z]
- 61. Deriving Optimal Treatment Timing for Adaptive Therapy [https://link.springer.com/article/10.1007/s11538-025-01525-y]
- 62. Advancing drug development with “Fit-for-Purpose†modeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12436579/]
- 63. A fractional-order model for optimizing combination therapy ... [https://www.nature.com/articles/s41598-024-66531-x]
- 64. How Advanced Math is Shaping the Future of Drug ... [https://engineering.virginia.edu/news-events/news/how-advanced-math-shaping-future-drug-development]
- 65. Benefit–risk assessment: the use of clinical utility index [https://www.semanticscholar.org/paper/Benefit%E2%80%93risk-assessment%3A-the-use-of-clinical-index-Ouellet/3eb353dba5944ab09dff641471a5e6091676a3be]
- 66. Comprehensive Guide to Clinical Trial Protocol Design & ... [https://www.clinicalleader.com/topic/clinical-trial-protocol-design-development]
- 67. Teaching troubleshooting skills to graduate students - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11407763/]
- 68. Troubleshooting Protocols - Biological Sciences: Summer ... [https://guides.library.cmu.edu/biologicalsciencesSRI/troubleshooting]
- 69. The Use of Mathematical Modeling in Preclinical Decision ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2022.860881/full]
- 70. Application of Mathematical Modeling and Computational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9268380/]
- 71. T3 + 3: 3 + 3 Design With Delayed Outcomes [https://pmc.ncbi.nlm.nih.gov/articles/PMC11602947/]
- 72. requiem for the 3 + 3 design for phase I trials [https://www.sciencedirect.com/science/article/pii/S0923753419317673]
- 73. Editorial: Model-informed drug development and precision ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2023.1224980/full]
- 74. Everything You Need to Know About BOIN Design in ... [https://www.quanticate.com/blog/boin-design-in-early-phase-clinical-trials]
- 75. Phase 1 Clinical Trial Designs Explained: BOIN, CRM ... [https://www.precisionformedicine.com/blog/phase-1-clinical-trial-designs-explained-boin-crm-blrm-modern-adaptive-strategies]
- 76. Model-Informed Dose Optimization of Pazopanib in Real- ... [https://link.springer.com/article/10.1007/s40262-025-01504-5]
- 77. Accuracy and Safety of Novel Designs for Phase I Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10237505/]
- 78. Digital twins for health: a scoping review [https://www.nature.com/articles/s41746-024-01073-0]
- 79. Frequently Asked Questions [https://www.digitaltwinconsortium.org/faq/]
- 80. Digital Twins for Personalized Medicine Require ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12365566/]
- 81. Digital twins in healthcare: a comprehensive review and ... [https://www.frontiersin.org/journals/digital-health/articles/10.3389/fdgth.2025.1633539/full]
- 82. Enhancing randomized clinical trials with digital twins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12494699/]
- 83. How can virtual patients transform the clinical trials process? [https://www.turing.ac.uk/blog/how-can-virtual-patients-transform-clinical-trials-process]
- 84. Designing combination therapies for cancer treatment [https://pmc.ncbi.nlm.nih.gov/articles/PMC11063284/]
- 85. Mathematical Optimization of Combination Drug Regimens [https://systemsmedicine.pulmonary.medicine.ufl.edu/more-about-uf-lsm/research/lsm-researcher-profiles/helen-moore-ph-d/determining-optimal-combination-regimens-for-patients-with-multiple-myeloma/]
- 86. Digital Twins in Healthcare: A Practical Guide for Clinics [https://holisticare.io/blog/digital-twins-in-healthcare/]
- 87. Virtonomy.io - Data driven clinical trials on virtual patients [https://www.virtonomy.io/]
- 88. Development of Cancer Drugs for Use in Novel Combination [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-cancer-drugs-use-novel-combination-determining-contribution-individual-drugs-effects]
- 89. Regulatory Focus – Stakeholders seek clarity from FDA on ... [https://friendsofcancerresearch.org/news/regulatory-focus-stakeholders-seek-clarity-from-fda-on-cancer-drug-combo-guidance/]